巨噬细胞USP9X通过K27连接去泛素化STAT1限制Oncostatin M产生减轻结肠炎
Macrophage USP9X Attenuates Colitis by Restricting Oncostatin M Production via K27-linked Deubiquitination of STAT1
Cellular & Molecular Immunology
PMID: 42209766 [IF=19.8]
来源:IBD Daily
泛素化作为一种关键的翻译后修饰,通过E3泛素连接酶和去泛素化酶 (DUBs) 的动态平衡,调控免疫信号传导、细胞因子产生和炎症消退。近期研究已揭示多种DUBs在肠道炎症中发挥重要调控作用。USP9X是一种多功能去泛素化酶,在肿瘤发生、免疫调节和代谢稳态中均有重要功能。然而,USP9X在肠道巨噬细胞生物学及IBD中的功能尚不明确。
本研究揭示了USP9X作为肠道炎症关键负调控因子的新功能:USP9X通过去除STAT1蛋白K544位点上的K27连接多聚泛素链,限制STAT1的核转位和转录活性,进而抑制下游Oncostatin M (OSM) 的产生。该研究为IBD的发病机制提供了新的翻译后修饰调控视角,并提示USP9X-STAT1-OSM轴可能是IBD的潜在治疗靶点。
研究者首先通过分析公共数据库中的转录组数据及独立临床队列,评估IBD患者肠道巨噬细胞中USP9X的表达变化及其与疾病严重程度的相关性。利用LysM-Cre系统构建髓系特异性Usp9x敲除小鼠 (Usp9xΔmye),分别在DSS和TNBS诱导的结肠炎模型中评估表型。通过RNA-seq、泛素化蛋白质组学、Co-IP、免疫荧光、PLA、CUT&Tag-seq、ChIP-qPCR等多种技术系统解析USP9X调控STAT1的分子机制。利用STAT1 K544R突变体及CRISPR/Cas9技术验证K544泛素化位点的功能。通过OSM中和抗体实验验证OSM在USP9X缺陷所致结肠炎中的关键效应作用。最后,采用双AAV9载体系统经骨髓腔内注射实现巨噬细胞中Usp9x的过表达,验证其治疗潜力。
1. IBD患者肠道巨噬细胞中USP9X表达显著降低,与疾病严重程度呈负相关
通过对两个大样本公共数据集 (GSE66407和GSE73661) 的分析,研究者发现IBD患者肠道黏膜活检组织中USP9X转录水平显著降低 (图1A-B)。在GSE73661数据集中,USP9X mRNA水平与Mayo内镜评分呈负相关 (图1C),提示USP9X表达降低与疾病严重程度增加相关。在来自本中心的独立UC验证队列中,USP9X转录水平与Mayo总评分及血沉 (ESR) 均呈显著负相关 (图1D-E)。
单细胞RNA测序数据 (GSE214695) 进一步揭示,USP9X在UC患者单核/巨噬细胞中的表达特异性降低 (图1F-G)。值得注意的是,在未成熟单核细胞 (CD14hiCD11bhiCCR2+CX3CR1+) 中,USP9X表达下降尤为显著,同时伴随促炎因子TNFα和IL6的表达升高 (图1I),提示USP9X可能在维持单核细胞正常成熟和抑制促炎活化中发挥重要作用。免疫荧光染色确认UC患者结肠CD68+巨噬细胞中USP9X蛋白表达显著低于健康对照 (图1J),DSS诱导的结肠炎小鼠结肠F4/80+巨噬细胞中也观察到类似降低。
体外实验表明,IFN-γ和LPS刺激可时间和剂量依赖性地下调BMDMs和HMDMs中USP9X的表达 (图1K)。用siRNA敲低RAW264.7细胞中的Usp9x后,巨噬细胞迁移和吞噬能力增强,促炎因子 (Tnfα、Il1β、Il6) 及M1极化标志物 (Nos2、Cd80) 表达上调,趋化因子Cxcl2升高,而M2标志物Cd206下调,表明Usp9x敲低促进巨噬细胞向促炎表型极化。
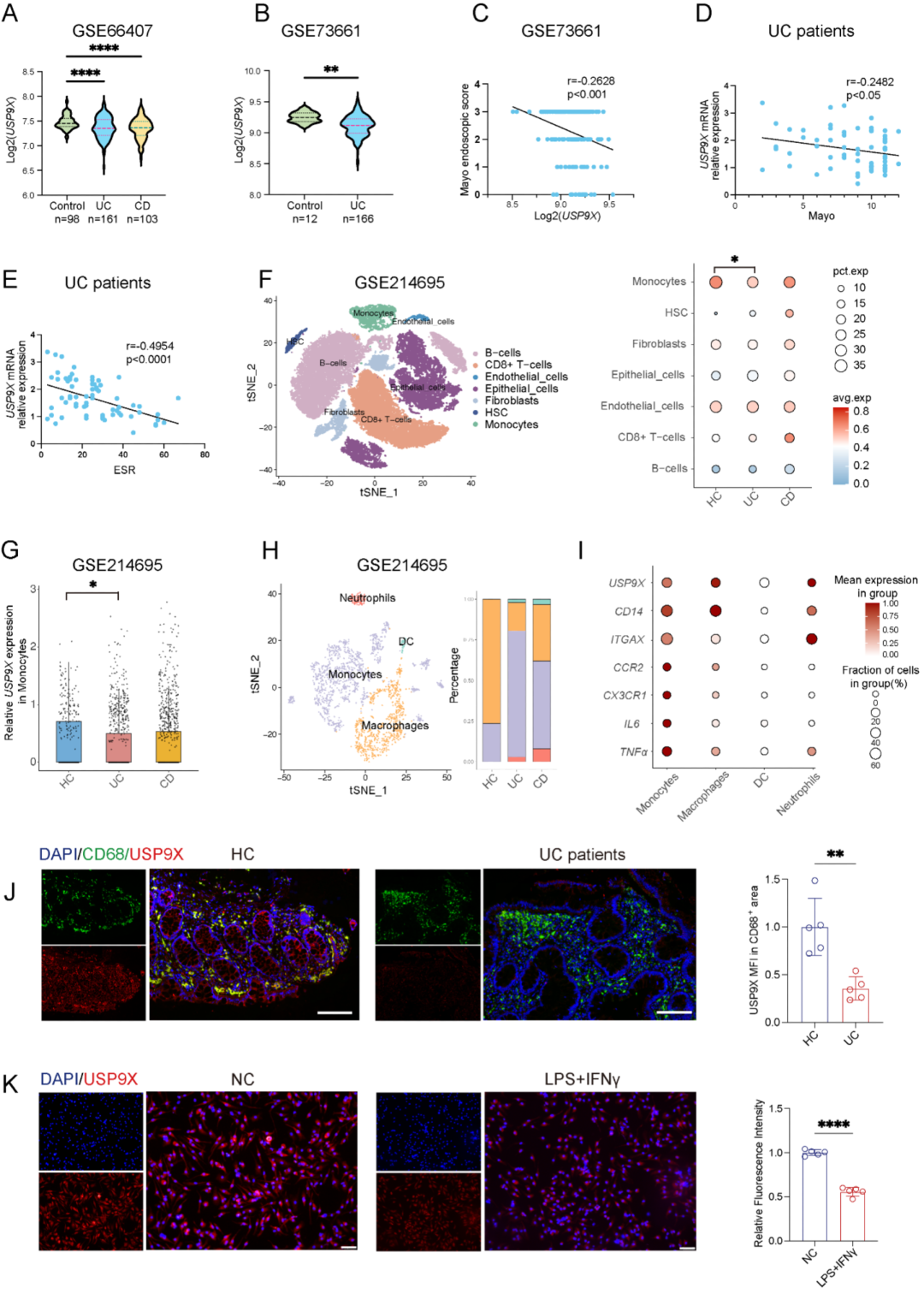
图1. USP9X在IBD患者中表达降低并与疾病严重度负相关。
2. 髓系特异性敲除USP9X促进炎性髓系细胞积累并加重实验性结肠炎
为探究USP9X在巨噬细胞中的体内功能,研究者构建了髓系特异性Usp9x敲除小鼠 (Usp9xΔmye)。在2.5% DSS诱导的急性结肠炎模型中,Usp9xΔmye小鼠较同窝对照出现更严重的结肠炎表现:结肠长度显著缩短、体重下降更明显、疾病活动指数 (DAI) 升高、组织病理学评分恶化 (图2A-D)。PAS染色和MUC2免疫组化显示杯状细胞数量减少,血清TNF-α、IL-1β、IL-6水平显著升高。在TNBS诱导的结肠炎模型和慢性DSS模型中得到了一致的结果验证。
流式细胞术分析固有层单个核细胞 (LPMCs) 发现,Usp9x缺失重塑了髓系细胞格局:Usp9xΔmye小鼠DSS处理后固有层中CD11b+Ly6C+单核细胞和CD11b+Ly6G+中性粒细胞比例显著增加 (图2E-F)。进一步通过单核-巨噬细胞成熟分群分析 (P1-P4,基于Ly6C和MHC-II表达),发现Usp9xΔmye小鼠中P1 (Ly6C+MHCII-) 未成熟单核细胞和P2 (Ly6C+MHCII+) 过渡态细胞显著积累,而P3/P4 (Ly6C-MHCII+) 成熟巨噬细胞明显减少 (图2G),表明USP9X缺失导致单核-巨噬细胞成熟受阻。
免疫荧光染色证实DSS处理后Usp9xΔmye小鼠结肠固有层F4/80+iNOS+M1型巨噬细胞增多 (图2H),而F4/80+CD206+M2型巨噬细胞比例无显著变化。重要的是,巨噬细胞清除实验 (
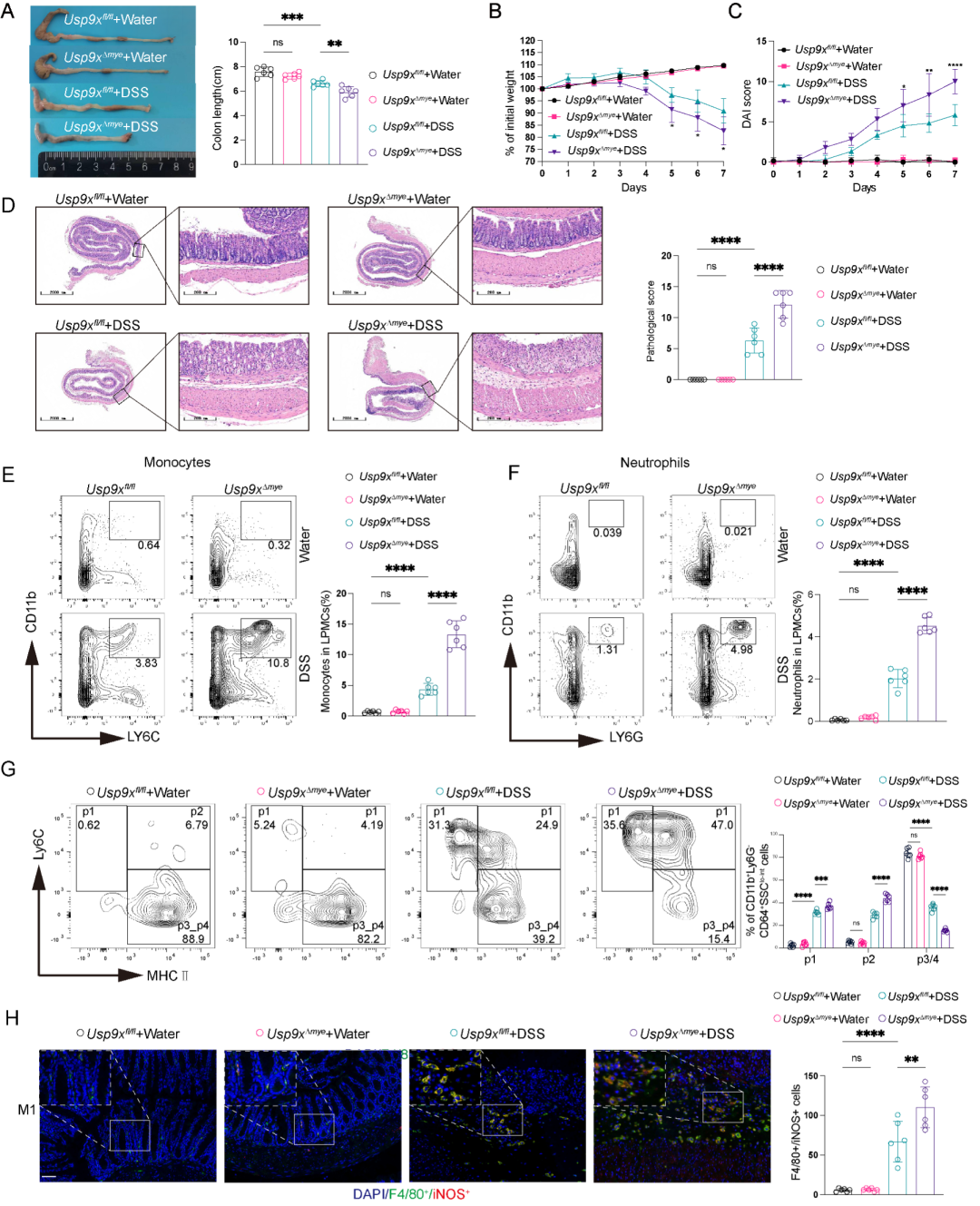
图2. 髓系特异性Usp9x缺失加重DSS诱导的结肠炎并增强炎性髓系反应。
3. USP9X缺失促进巨噬细胞促炎活化
为在转录组水平系统评估USP9X缺失对巨噬细胞的影响,研究者对Usp9xfl/fl和Usp9xΔmye小鼠的BMDMs进行RNA-seq分析 (图3A-C)。KEGG通路富集分析显示,Usp9x缺陷BMDMs中上调的基因显著富集于免疫相关通路,特别是"细胞因子-细胞因子受体相互作用"通路等 (图3D)。GSEA分析进一步证实TNF信号通路和细胞因子-细胞因子受体相互作用在Usp9xΔmyeBMDMs中显著富集 (图3E-F),揭示USP9X缺失导致巨噬细胞整体炎症性转录编程增强。体外功能实验表明,Usp9x缺陷BMDMs中促炎因子、趋化因子和M1标志物的mRNA表达显著上调 (图3G),迁移和吞噬能力明显增强 (图3H-I)。
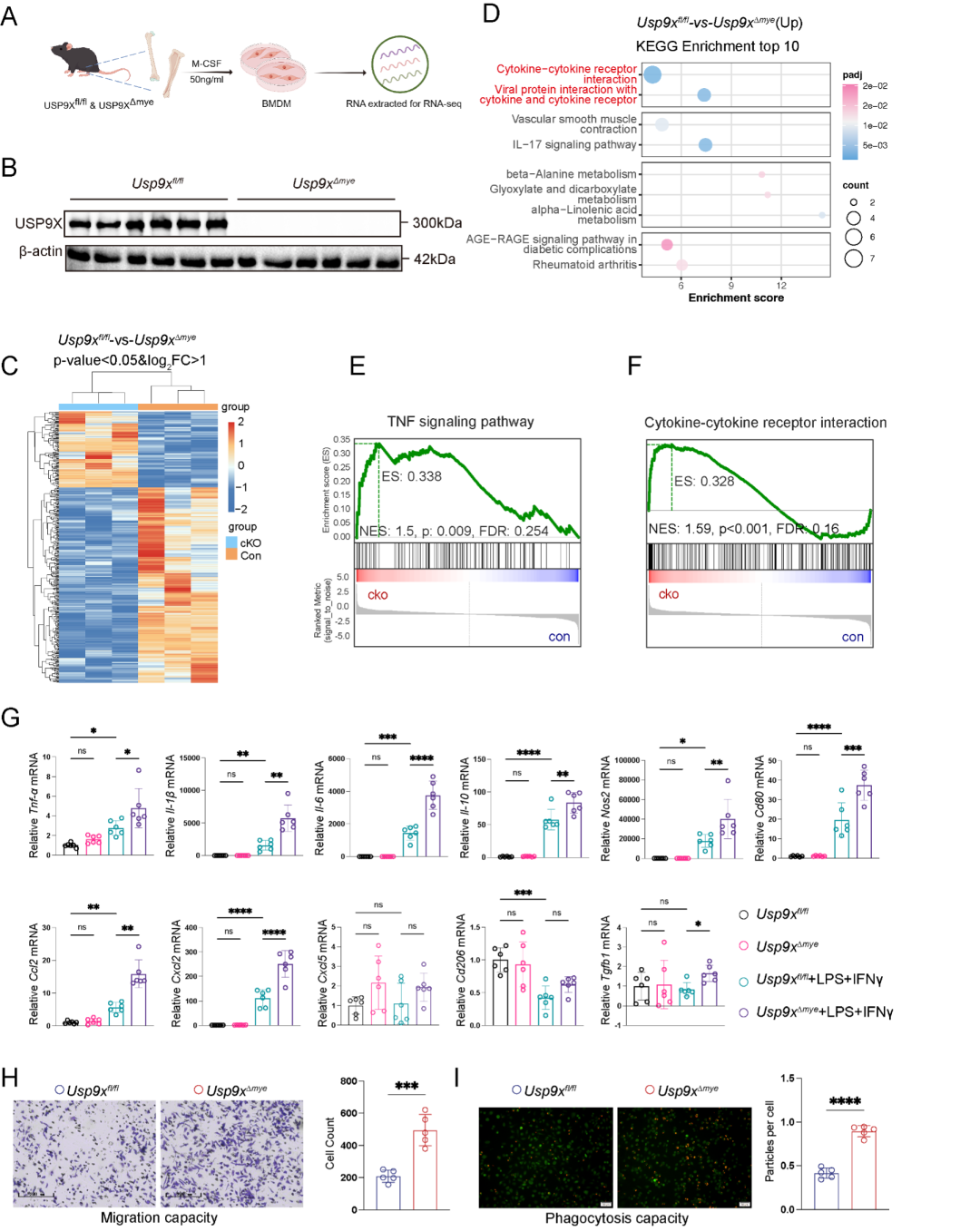
图3. USP9X缺失增强巨噬细胞促炎活化、迁移和吞噬能力。
4. USP9X直接结合STAT1并特异性去除K544位点的K27连接多聚泛素链
为揭示USP9X调控巨噬细胞炎症的分子底物,研究者采用了整合蛋白质组学策略。基于前期label-free泛素化蛋白质组学数据 (Usp9x敲低后132个泛素化位点上调≥2倍),结合FLAG-USP9X免疫沉淀-质谱鉴定出的592个USP9X互作蛋白,二者取交集获得24个候选直接底物 (图4A-D)。在其中,研究者聚焦于STAT1,一种调控巨噬细胞活化和炎症基因表达的关键转录因子。质谱分析提示Usp9x沉默后STAT1在K544位点的泛素化增加。
PLA和免疫荧光共定位实验在HMDMs中确认了USP9X与STAT1的胞内相互作用 (图4E-F)。内源性和外源性Co-IP实验在BMDMs及HEK293T细胞中均验证了两者的物理结合 (图4G-H)。通过USP9X截短突变体分析,定位STAT1结合区域为USP9X的C1片段 (含催化结构域)。
泛素化实验表明,USP9X过表达显著降低STAT1多聚泛素化水平,而DUB抑制剂WP1130可阻断此效应,证明STAT1的去泛素化依赖USP9X的催化活性 (图4I-J)。利用仅保留单个
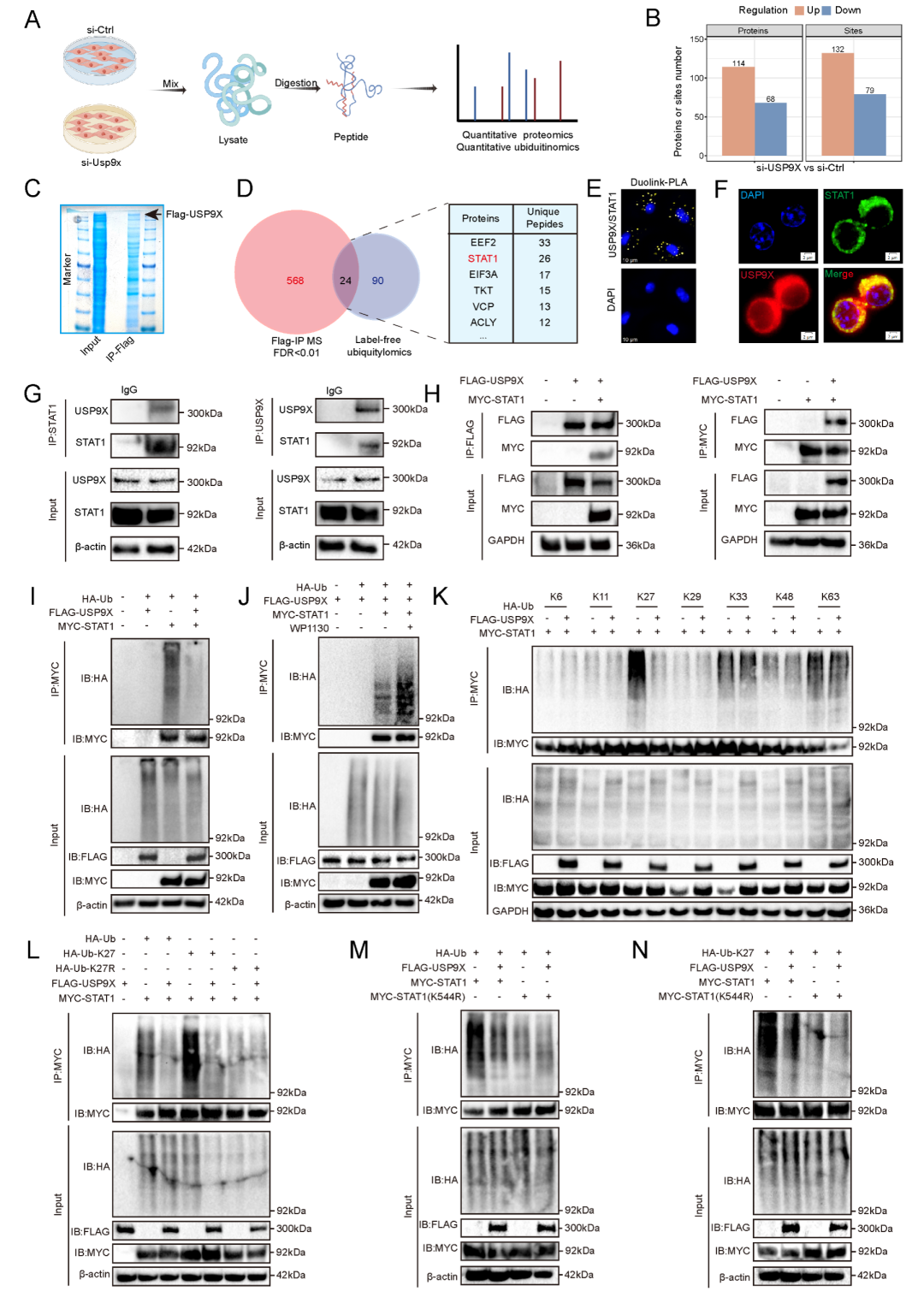
图4. USP9X与STAT1直接互作并特异性去除K544位点的K27连接多聚泛素链。
5. STAT1 K544位点泛素化是USP9X调控巨噬细胞炎症所必需
为验证STAT1 K544泛素化在USP9X调控巨噬细胞活化中的功能意义,研究者利用CRISPR/Cas9技术构建了Stat1敲除 (STAT1-/-) RAW264.7细胞,并通过慢病毒系统分别回补野生型STAT1 (STAT1WT) 或K544R突变体(STAT1K544R) (图5A-B)。结果显示,Usp9x敲低显著增强STAT1+/+巨噬细胞的迁移和吞噬能力,但STAT1-/-细胞中这些效应完全消失 (图5C-D)。RT-qPCR分析进一步证明,Usp9x敲低诱导的促炎基因 (Tnfα、Il-1β、Il-6、Nos2)上调完全依赖STAT1的存在 (图5E)。
在比较STAT1WT与STAT1K544R回补细胞时发现,Usp9x敲低在STAT1WT细胞中增强迁移和吞噬功能,但在STAT1K544R细胞中无此效应 (图5F-G)。STAT1K544R巨噬细胞在LPS/IFN-γ刺激后的炎症基因 (Tnfα、Il-6、Ccl2、Cd80) 表达显著减弱 (图5H)。体内实验中,利用F4/80启动子驱动的AAV载体实现巨噬细胞特异性STAT1WT或STAT1K544R表达,STAT1K544R组小鼠DSS结肠炎严重程度显著减轻,临床和组织学参数均有改善。这些结果证明STAT1 K544位点对于USP9X调控STAT1依赖性炎症反应至关重要。
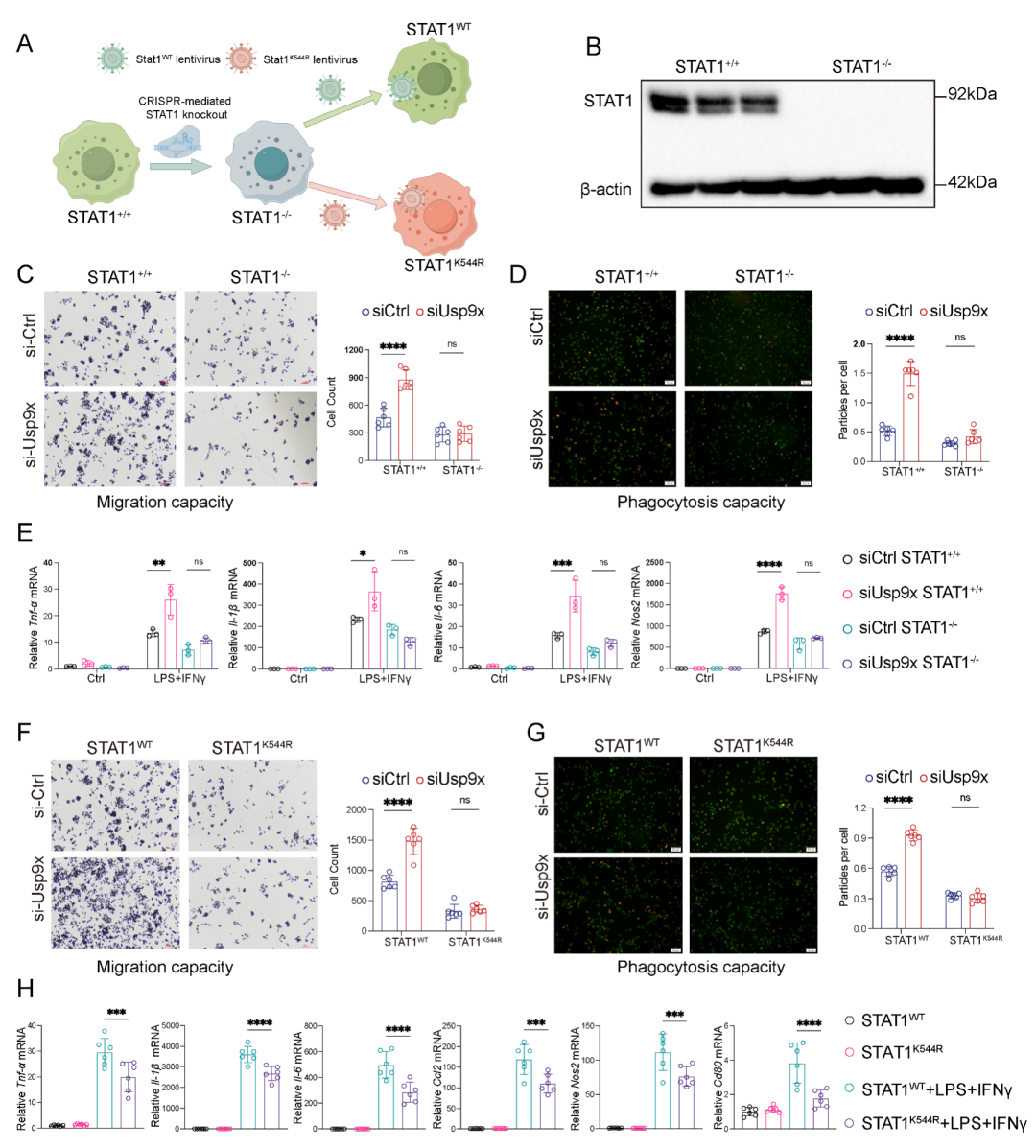
图5. STAT1 K544位点突变降低巨噬细胞促炎活化。
6. USP9X缺失增强STAT1核转位和染色质结合,驱动OSM表达
在确定USP9X介导STAT1 K544去泛素化后,研究者系统探究了这一修饰对STAT1功能的影响。蛋白质半衰期分析 (CHX追踪实验) 显示USP9X缺失不影响STAT1蛋白稳定性。JAK1-STAT1相互作用和STAT1 Y701磷酸化动力学在Usp9xfl/fl和Usp9xΔmyeBMDMs间无显著差异,表明上游JAK-STAT信号起始传导保持完整。
然而,尽管磷酸化水平相当,Usp9x缺陷BMDMs中STAT1的核转位显著加速和增强:免疫荧光显示核STAT1水平更早达到峰值 (0.5h vs. 1h) 且峰值水平更高 (图6A-B)。核蛋白免疫印迹进一步验证了STAT1在Usp9xΔmyeBMDMs核组分中的富集 (图6C)。3小时后两种基因型均表现出相似的核输出动力学,提示K27泛素化特异性促进核输入步骤而非影响核滞留。重要的是,STAT1 K544R突变体无论USP9X状态如何,其核转位均受损,表明K27连接泛素化是STAT1高效核输入的必要条件。
为探究增强的核定位是否转化为染色质结合的增加,研究者对DSS处理后Usp9xfl/fl和Usp9xΔmye小鼠固有层F4/80+CD11b+巨噬细胞进行CUT&Tag-seq分析 (图6D)。STAT1结合峰主要富集于启动子区域,Usp9x缺陷组在转录起始位点 (TSS) 周围的信号强度整体增加 (图6E-G)。
整合CUT&Tag-seq和RNA-seq数据,鉴定出92个在Usp9x缺陷巨噬细胞中STAT1启动子结合增加且转录上调的基因,其中9个落在GO“炎症反应”条目中 (图6H)。在这些候选基因中,Oncostatin M (OSM),一种IL-6家族细胞因子,引起了研究者的特别关注。OSM已在Nature Medicine等顶刊中被证实是IBD的重要生物标志物,其高表达与抗TNF治疗失败密切相关。
基因组浏览器可视化显示Usp9x缺陷巨噬细胞中STAT1在Osm启动子区域的富集增强 (图6I)。ChIP-qPCR针对Osm启动子三个区域 (P1-P3) 验证了STAT1结合增加 (图6J)。重要的是,Usp9x敲低仅在STAT1WT背景下诱导Osm表达,而在STAT1K544R细胞中无效,强调了K544位点对USP9X介导的OSM调控的必要性。UC患者结肠组织免疫荧光染色直观显示,与健康对照相比,USP9X表达降低而OSM表达升高 (图6K)。这些结果表明OSM是STAT1的直接转录靶基因,是USP9X-STAT1信号轴的关键下游效应分子。
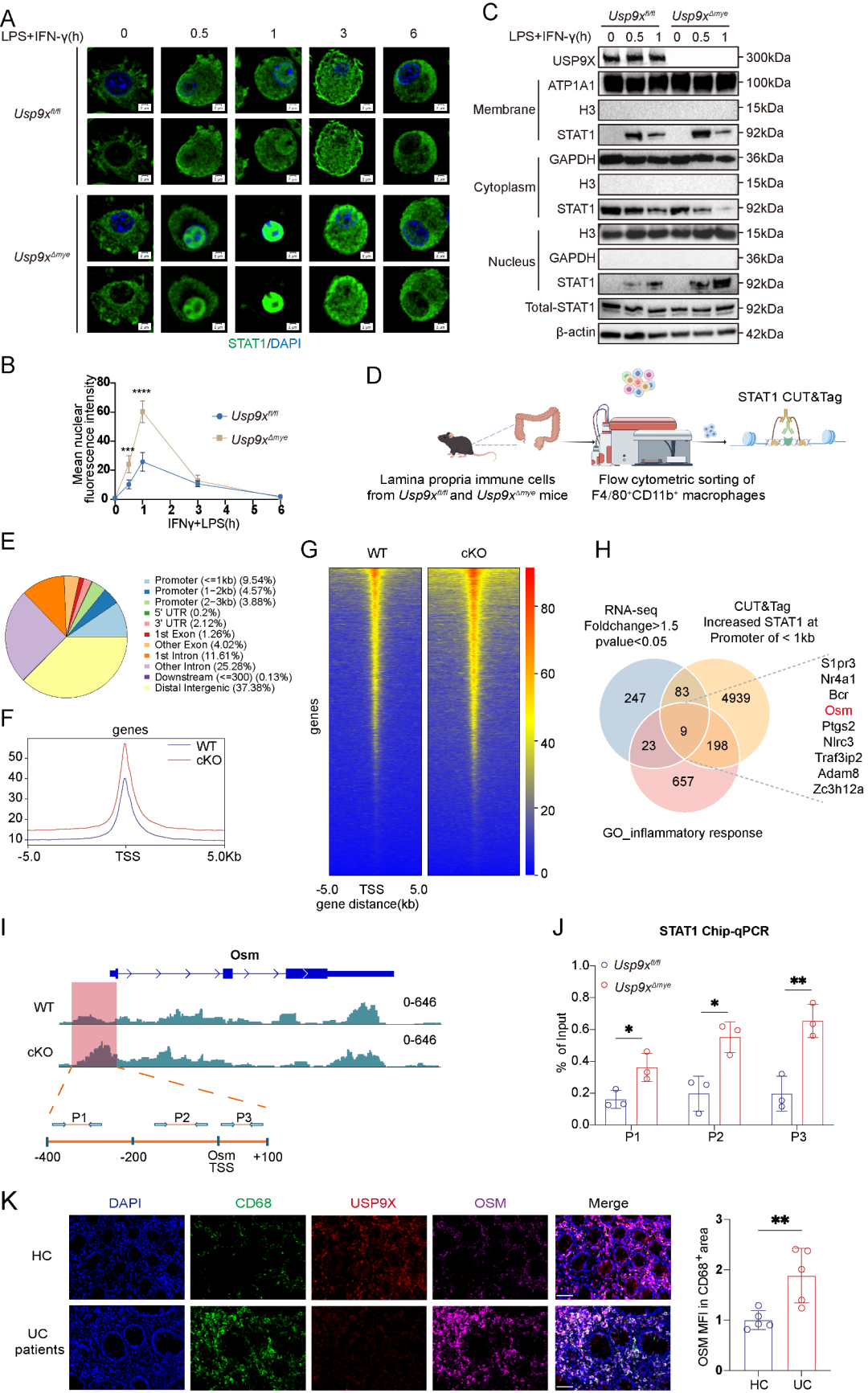
图6. USP9X缺失增强STAT1核转位和染色质结合促进OSM表达。
7. 中和OSM可逆转USP9X缺失导致的结肠炎加重
为在体内验证OSM是否为USP9X-STAT1信号轴的关键下游效应分子,研究者对DSS处理的Usp9xfl/fl和Usp9xΔmye小鼠给予OSM中和抗体 (于DSS给药后第3、5天腹腔注射) (图7A)。未经抗体治疗时,Usp9xΔmye小鼠较对照组表现出更严重的结肠炎。OSM阻断显著改善了两种基因型小鼠的体重下降、DAI评分、结肠长度和组织病理学评分,伴随黏膜结构恢复、杯状细胞增加 (MUC2和PAS染色) (图7A-E),且最重要的是完全消除了Usp9xfl/fl和Usp9xΔmye小鼠之间结肠炎严重程度的基因型差异。
机制层面,OSM中和抑制了结肠组织中由USP9X缺失引起的STAT3磷酸化增强,这与近期Nature Immunology报道的“OSM在结肠炎中激活肠上皮STAT3信号、增加趋化因子产生和免疫细胞募集”的机制一致。流式细胞术显示,OSM阻断减少了Usp9x缺陷结肠中CD11b+Ly6C+单核细胞和CD11b+Ly6G+中性粒细胞的积累 (图7F-G),并恢复了被破坏的单核-巨噬细胞成熟进程 (P1/P2减少,P3/P4增加) (图7H)。这些结果确立了OSM作为USP9X下游功能性效应因子、通过增强促炎性髓系活化驱动肠道炎症的核心地位。鉴于OSM高表达预测抗TNF治疗失败,USP9X-STAT1-OSM轴的阐明为克服IBD治疗抵抗提供了新策略。
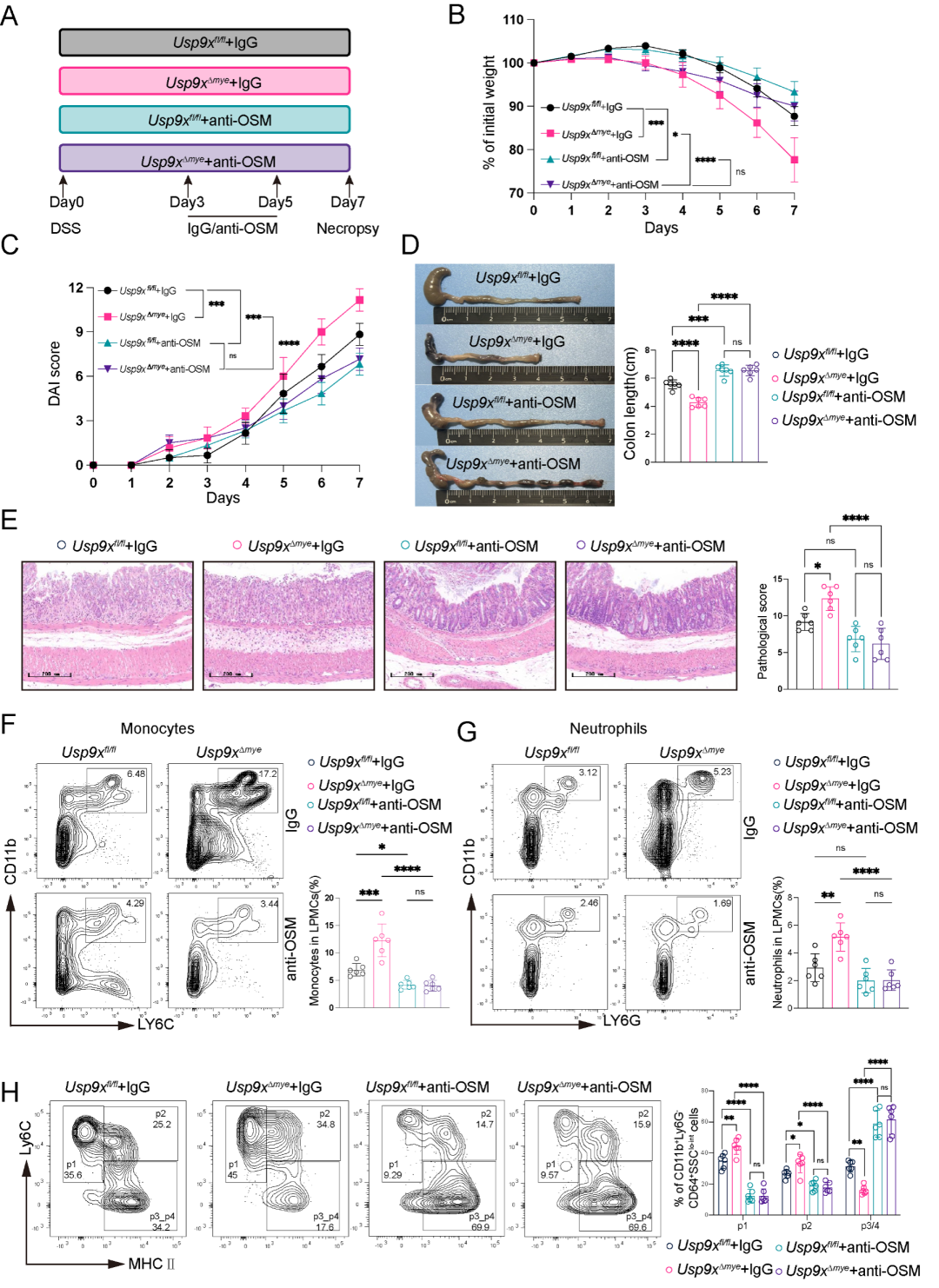
图7. OSM中和抗体逆转USP9X缺陷所致结肠炎加重。
8. 髓系细胞中过表达USP9X可减轻DSS诱导的结肠炎
为探索USP9X的治疗潜力,研究者采用基于REVeRT技术的双AAV9载体系统。由于Usp9x编码序列 (约7.5 kb) 超出单个AAV载体的包装容量,研究者将基因拆分为5'端和3'端片段,分别包装于两个AAV9载体中,通过mRNA反式剪接 (trans-splicing) 在靶细胞中重建全长Usp9x转录本和蛋白。通过骨髓腔内注射将双AAV9载体递送至造血祖细胞及骨髓来源的髓系细胞 (图8A-C)。
注射一个月后,BMDMs中USP9X蛋白表达得到有效恢复 (图8B)。在随后进行的2.5% DSS结肠炎模型中,与AAV-对照小鼠相比,AAV-Usp9xOE小鼠表现出全面的保护效应:结肠长度得到保留 (图8D)、体重下降减轻 (图8E)、DAI评分降低 (图8F),组织学分析显示黏膜损伤减轻、杯状细胞数量增加 (MUC2免疫组化和PAS染色) (图8G)。血清TNF-α、IL-1β和IL-6水平显著下降 (图8H)。免疫沉淀实验证实USP9X过表达在体内降低了STAT1泛素化水平 (图8I),直接证明了USP9X-STAT1去泛素化轴在体内的功能相关性。这些结果展示了靶向USP9X-STAT1轴治疗IBD的可行性和转化潜力。
图8. AAV9介导的USP9X过表达减轻小鼠结肠炎。
本研究首次揭示了USP9X作为肠道炎症关键负调控因子的新功能,系统阐明了“USP9X-STAT1-OSM”这一全新的翻译后修饰调控轴:USP9X通过去除STAT1 K544位点的K27连接多聚泛素链,限制STAT1核转位和染色质结合,抑制下游OSM转录,从而维持肠道巨噬细胞正常成熟和免疫稳态。USP9X缺失则导致STAT1 K27泛素化积累、异常核转位增强、OSM过度产生,最终驱动促炎性髓系细胞激活和肠道炎症加剧。
该研究具有多重创新意义:第一,在功能层面将K27连接泛素化与STAT1核转位调控联系起来,扩展了“泛素编码 (ubiquitin code)”概念,证明不同泛素链拓扑结构可赋予转录因子以环境特异性的基因激活模式;第二,发现STAT1是OSM此前未知的直接转录激活因子,填补了OSM转录调控领域的重要空白;第三,鉴于OSM已被证实为IBD抗TNF治疗失败的强预测标志物,USP9X-STAT1-OSM轴的阐明为克服IBD治疗抵抗提供了全新的干预靶点。特别值得关注的是,USP9X与OSM在患者组织中的反向表达关系提示,USP9X表达水平可能用于筛选抗OSM或JAK靶向治疗的获益人群,为IBD的精准治疗提供生物标志物依据。
论文发表于Cell Mol Immunol杂志,得到天津医科大学总医院消化中心主任
 我要投稿
我要投稿














{kind=link}