
编者按
角层下脓疱性皮病(Subcorneal Pustular Dermatosis, SPD)又称 Sneddon-Wilkinson 病,是一种罕见的嗜中性皮病。本刊报道一例继发于系统性


主诉
患者,女性,40余岁,因躯干瘙痒性
现病史
患者于5天前无明显诱因于躯干出现红色斑片,迅速扩展至四肢近端,伴有明显瘙痒。皮疹表现为多发性松弛性脓疱,内含清亮与浑浊液体,呈特征性上清下脓样脓疱(half-half pustule)形态。病程中无
既往史
确诊B细胞
体格检查
-躯干及四肢近端可见 红斑性斑片,其上分布大量松弛性分层脓疱,部分脓疱融合形成环状或匐行性斑块(图A、B)。
-脓疱内液体呈水平分层:上半部清亮液体,下半部脓性液体。
-无黏膜受累,面部、手掌、足底未见
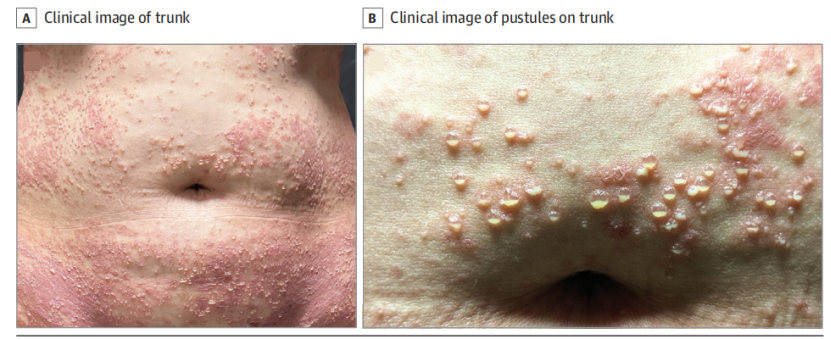
图1A:躯干红斑基础上可见大量特征性上下分层状脓疱。
图1B:脓疱融合形成环状或匐行性斑块,部分脓疱破溃并形成浅表痂皮。
辅助检查
-皮肤组织病理:角层下中性粒细胞性脓疱。
-直接免疫荧光:阴性。
-特殊染色:抗酸染色、PAS 染色、GMS 染色均未检出病原微生物。
-组织培养:细菌、真菌、分枝杆菌培养均阴性。


结合临床表现、病理特征及辅助检查结果,诊断为角层下脓疱性皮病(SPD,Sneddon-Wilkinson 病)。


鉴于患者存在血液学相关风险,且G6PD状态未知,未使用一线药物
皮损1周内完全消退,随访1年无复发。


SPD是一种罕见的慢性嗜中性皮病,好发于中年女性,典型表现为复发性无菌性脓疱,因重力作用形成水平分层(上清下脓)。皮损好发于屈侧及间擦部位,常融合成环状或匐行性斑块。
本例患者无黏膜受累,面、掌、足底不受累,符合典型SPD特征。值得注意的是,患者在皮损出现前3周曾接受系统性G-CSF治疗。已有文献报道G-CSF注射部位可诱发局限性的SPD样反应,而本例为系统性G-CSF暴露后出现的广泛性SPD,提示G-CSF可能在易感个体中诱导全身性中性粒细胞活化,扩展了细胞因子相关嗜中性皮病的疾病谱。
SPD发病机制尚未完全阐明,可能与中性粒细胞趋化因子及免疫失调相关。SPD可与多种系统性疾病相关,包括:IgA副蛋白血症、多发性骨髓瘤、类风湿


需与泛发性脓疱型
SPD鉴别要点:慢性复发性病程、无明显全身症状、病理示角层下无菌性脓疱、直接免疫荧光阴性。


-一线治疗:口服氨苯砜。
-二线/难治性病例:系统
-合并


对于接受G-CSF 治疗的血液肿瘤患者,需警惕包括 SPD 在内的嗜中性皮病。SPD具有特征性临床与病理表现,早期识别与规范治疗预后良好,长期系统随访以筛查潜在合并疾病至关重要。
参考文献:
医脉通是专业的在线医生平台,“感知世界医学脉搏,助力中国临床决策”是平台的使命。医脉通旗下拥有「临床指南」「用药参考」「医学文献王」「医知源」「e研通」「e脉播」等系列产品,全面满足医学工作者临床决策、获取新知及提升科研效率等方面的需求。

 我要投稿
我要投稿














{kind=link}