抽丝剥茧:“铜蓝蛋白消失”谜案,终现“铁证如山”丨病例分享
2026-01-20
近期,中山三院感染性疾病科在Gastroenterology(IF:25.9)杂志在线报道了一例罕见遗传代谢性疾病。该患者在铁与铜的指标之间出现看似矛盾的表现,最终通过严密的逻辑推理,层层递进,揭开谜底。以下为本病例的深度解析与分享。
患者45岁男性,因“失眠20个月,腹胀不适2个月”入院。用药史及家族史无特殊。20个月前患者因“睡眠差、失眠”在当地医院诊断为“糖尿病”,发现血清铁蛋白2233 ng/mL,铜蓝蛋白< 0.024 g/L,肝脏CT值85.62 HU(图A)。2月前出现腹胀不适,查铁蛋白19710 ng/mL,ALT 301 U/L,AST 190 U/L,当地医院给予中药及护肝药物治疗,肝功能有所好转,病因未明,入我院行进一步诊治。
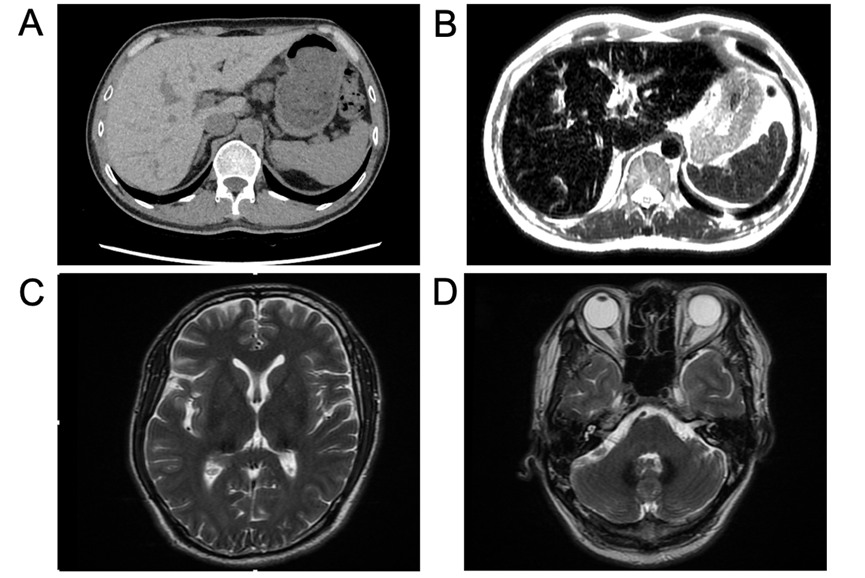
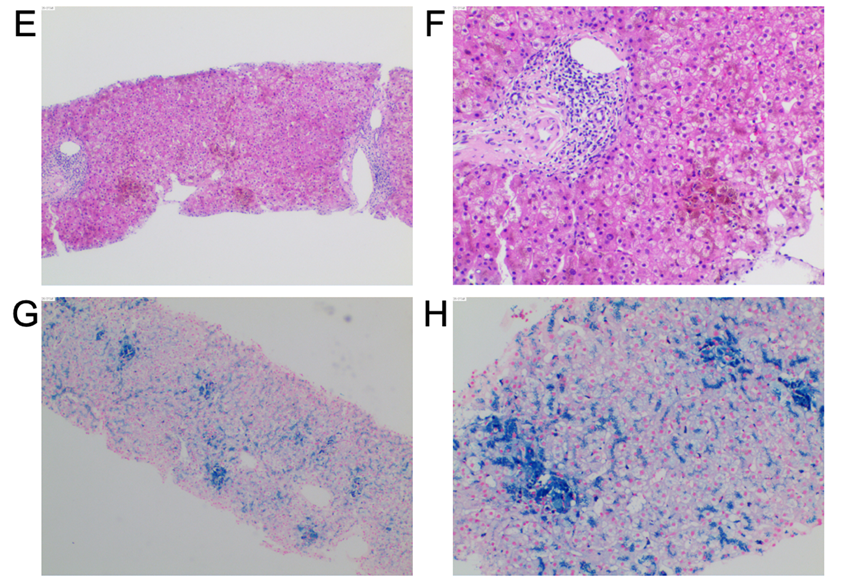
查体:胸前毛细血管扩张(+),无肝掌及蜘蛛痣。实验室检:血红蛋白118 g/L,白细胞和血小板计数正常,网织红细胞3.09%。肝功能及凝血功能基本正常,血糖 15.27 mmol/L,总胆固醇5.19 mmol/L,血清铁14.4 μmol/L,转铁蛋白饱和度45.1%,糖化血红蛋白6.2%。肝炎病毒标志物、自身免疫性抗体检查均为阴性。彩超肝胆脾胰未见明显异常。眼科:视网膜平伏,未见出血或渗出。腹部MRI-T2相显示肝脏、胰腺及脾脏明显低信号(图B)。头颅MRI显示双侧丘脑、基底节区和齿状核存在对称性斑片状短T2信号(图C、D)。骨髓穿刺涂片、组织病理学活检和染色体核型分析均未发现异常。肝脏穿刺活检显示慢性轻度炎症性肝损伤(G1S1)改变,肝细胞和库普弗细胞内有显著的含铁血黄素沉积,普鲁士蓝染色呈阳性(图E-H)。
患者低铜蓝蛋白、肝及脑同时受累,肝脏CT值显著增高(提示金属高密物质沉积),影像与检验结果让临床医生很容易想到肝豆状核变性,但三个矛盾点却让这个诊断陡然生疑:1.患者极低铜蓝蛋白而24小时尿铜却正常?肝脏病理亦未见铜沉积;2.肝脏、胰腺及脾脏MRI-T2明显低信号提示铁过载,这也与肝脏病理显著的含铁血黄素沉积一致,这些结果指向血色病,但是血清铁及转铁饱和度却是正常?3. MRI-T2“黑肝黑脾”、贫血,指向红细胞疾病继发性血色病,但是影像却无脾大。
这些看似不可能共存的极端指标,临床可以排除WD,却指向了一种罕见的铁代谢性疾病。肝脏重度铁过载的影像证据确凿,但血清铁及转铁蛋白饱和度却出奇地正常——这强烈暗示,铁代谢紊乱的根源并非肠道对铁的吸收失控。真正的线索,恰恰藏在这一系列矛盾组合之中:极低的铜蓝蛋白、头颅MRI揭示的特定脑区铁沉积信号……这些碎片拼凑起来,高度指向一个更为罕见的真相——一种由CP基因突变导致铜蓝蛋白功能完全丧失,进而引发的铁代谢异常:无铜蓝蛋白血症(Aceruloplasminemia, ACP)。
1、临床表现提供初始线索:从睡眠差、失眠等神经系统症状前兆,到腹胀、腹部不适等消化系统症状,再到已明确的糖尿病和轻度贫血,构成了多系统受累的复杂表现。
2、实验室检查揭示核心矛盾:(1)铜蓝蛋白极度低下(< 0.024 g/L),24小时尿铜排泄量却正常(15.9 μg/24h);(2)血清铁蛋白飙升至19710 ng/mL,转铁蛋白饱和度却保持正常(45.1%),血清铁 14.4 μmol/L;(3)血糖 15.27 mmol/L,糖化血红蛋白6.2%,糖尿病的诊断明确。
3、影像学检查获铁沉积关键证据:腹部CT与MRI清晰显示肝脏、胰腺等器官重度铁过载的特征,而脾脏铁沉积轻;头颅MRI发现双侧丘脑、基底节区等关键脑区的对称性铁沉积,这是ACP重要的临床表现。
4、病理检查显示铁过载直接证据:肝脏活检显示肝小叶结构存在,部分肝细胞水样变性,可见点、灶状坏死,肝细胞胞浆见较多含铁血黄素沉积;个别汇管区扩大,少量淋巴细胞、少许浆细胞浸润,未见确切界面炎及桥接坏死,小胆管形态及分布尚可,纤维组织轻度增生,符合慢性炎性肝损伤(G1S1)改变伴明显铁过载。虽同样显示显著的铁沉积和慢性轻度炎症,但其铁主要沉积于巨噬细胞内,没有明显的梯度递减,这与原发性血色病典型的、以肝实质细胞为主且呈梯度分布的沉积模式有所不同。
5、基因测序锁定铁证:在患者CP基因的c.1864+5G>A位点上发现了纯合突变。这份分子层面的诊断报告,为整个扑朔迷离的病例画上了确切的句号,也清晰地揭示了“消失的铜蓝蛋白”背后隐藏的疾病本质。
患者长期服用二甲双胍和恩格列净以控制血糖。随后开始口服地拉罗司,剂量为10-15毫克/公斤/天。患者对该治疗方案耐受良好,日常生活未受影响。
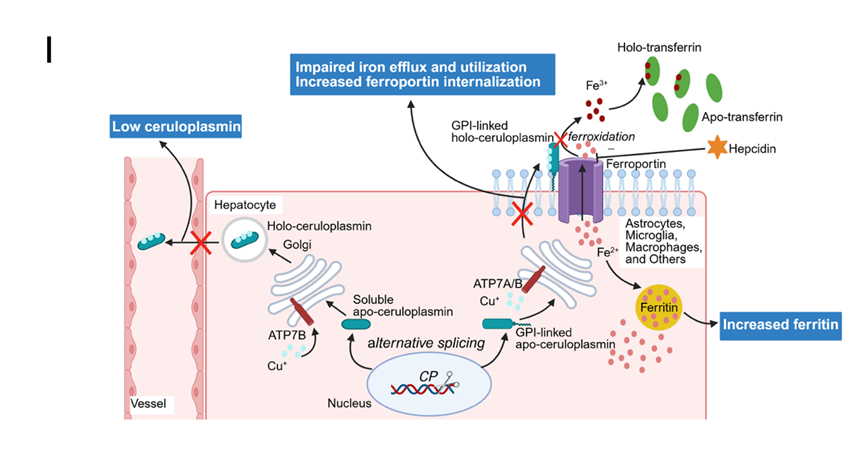
ACP是由定位于3q22-25的血清铜蓝蛋白基因(CP)突变所引起的一种极为罕见的常染色体隐性遗传疾病。据最新流行病学研究,ACP的患病率可达十万分之12.6。CP基因突变类型主要为错义突变、移码突变,其次为剪接突变、无义突变,且纯合突变占多数。致病基因CP编码铜蓝蛋白前体α2球蛋白,含有19个外显子和18个内含子。膜锚定的铜蓝蛋白为亚铁氧化酶,负责催化Fe2+氧化为Fe3+,在铁代谢中发挥着至关重要的作用。CP基因突变主要通过以下机制影响临床表现:① 可溶性CP的下降,出现血清铜蓝蛋白的显著下降;② 铁不能流出单核巨噬细胞,铁沉积在细胞内,进而引起骨髓利用铁障碍,出现缺铁性贫血及MRI-T2影像上“黑肝黑脾”的表现;③ 铁在细胞内储积,促进铁蛋白的合成,进而引起血清铁蛋白的显著增高;④ 铁流出障碍及Fe2+不能与转铁蛋白结合,导致血清铁及转铁蛋白饱和度不高。CP基因缺陷通过以上机制引起严重的铁代谢紊乱,导致Fe2+沉积在脑、肝、胰腺等造成多器官损害(图I)。
ACP平均发病年龄在50岁,病程长,典型临床表现为糖尿病、视网膜变性和神经退行性症状的“三联征”,伴有肝脏炎症,同时也可能引起消化功能的障碍,脂质代谢失调等。早期神经系统症状不明显时,可能仅表现为不明原因的轻度小细胞低色素贫血或实验室指标异常,具有高血清铁蛋白和铜蓝蛋白明显缺乏,然而,绝大多数患者确诊时已出现难以逆转的认知功能障碍,或共济失调等运动障碍。目前尚未有研究发现基因突变位点与临床表型存在明显关联性。
由于ACP相关的临床及影像学表现,容易被误诊为肝豆状核变性。当遇到以下特点时,应怀疑ACP:(1)铜蓝蛋白明显降低或低于检测值,血清铁蛋白明显升高,转铁蛋白饱和度正常或下降,而其他铜代谢相关指标正常;(2)腹部磁共振成像显示肝脏、胰腺重度铁过载,而脾脏铁沉积轻;(3)头颅磁共振显示丘脑、基底节区、齿状核等显示出明显的双侧对称性均匀低信号。
肝脏穿刺活检对于ACP诊断同样有重要意义。在肝脏铁沉积组织学分布模式中,原发性血色病主要表现为肝实质细胞型,铁沉积从1带到3带减少,而ACP表现以巨噬细胞内铁沉积为主,没有明显的递减梯度,且铜沉积为阴性(图E-H),但是很少导致肝硬化或肝衰竭等。
然而,ACP的确诊最终依靠基因诊断。患者CP基因的c.1864+5G>A位点上存在纯合突变,这个变异曾报道3例,突变会导致野生型供体剪接位点缺失,干扰CP剪接,激活隐匿的剪接位点,产生无功能的、被降解的mRNA,或者异常缩短的蛋白质,最终导致铜蓝蛋白显著减少或完全缺失。
目前对于ACP的治疗推荐使用去铁螯合治疗,目前临床使用铁螯合剂主要为驱铁胺DFO、去铁酮DFP及地拉罗司DFX。DFO 每日静脉给药用量为 25~60 mg/kg,DFP 每日用量为 70-75 mg/kg,DFX 每日用量为10~15 mg/kg,但铁螯合剂对于神经系统症状改善效果有限。其次为放血疗法或治疗性红细胞去除术,这需要提前评估患者能否耐受贫血。铁调素和CP替代疗法尚处于实验研究阶段。
本例诊治过程凸显了面对复杂代谢指标矛盾时,拓宽诊断思维、多学科协作与基因检测技术的重要性。对于疑似肝豆状核变性但存在矛盾点的病例,应警惕更罕见疾病的可能性,从而实现早期诊断与干预。
李新华
主任医师,医学博士,博导
专业特长:从事各种传染性疾病的临床诊治工作,擅长遗传代谢性肝病及各种罕见、疑难肝病的诊疗,如肝豆状核变性、卟啉病、血色病、胆汁淤积性肝病、自身免疫性肝病等。
学术成果:主持和参与了国家重点研发计划及国家自然科学基金项目等多项科学研究。研究方向主要集中在疑难罕见肝脏疾病的诊断、治疗和发病机制等方面。以第一作者或通讯作者在《Gastroenterology》、《J Hepatol》等杂志发表SCI论文40余篇,参与编写《肝豆状核变性诊疗指南(2022版》等多项指南、共识和标准。荣获广东省杰出青年医学人才、广东省青年五四奖章个人奖、中组部“第十批省市援疆工作优秀个人”并记功一次等荣誉。
许镇
副主任医师,医学博士,硕导
专业特长:感染性疾病专家,擅长病毒性肝炎、肝硬化、肝衰竭及疑难肝病、遗传代谢肝病的诊治。研究方向:慢乙肝临床治愈相关免疫机制、疑难肝病、I期临床研究。
成果简介:中山大学校级优秀教师。从事医教研工作20年,任国家住院医师规范化培训重点专业基地秘书。兼任广东省药学会肝脏病专家委员会秘书、广东省老年保健协会疑难肝病专业委员会副主任委员、中国医师协会感染科医师分会委员等学术职务,为广州市健康科普专家。
供稿:王睿欣
编辑:陈洁
审核:李小燕、李新华
审定:张晓红、林炳亮
Wang R, Chen J, Li X. Unexplained Coexisting Injury of the Liver, Pancreas, and Brain. Gastroenterology. 2026 Jan 5:S0016-5085(26)00003-X. doi: 10.1053/j.gastro.2025.12.031.
来源:中山三院感染性疾病科
医脉通是专业的在线医生平台,“感知世界医学脉搏,助力中国临床决策”是平台的使命。医脉通旗下拥有「临床指南」「用药参考」「医学文献王」「医知源」「e研通」「e脉播」等系列产品,全面满足医学工作者临床决策、获取新知及提升科研效率等方面的需求。
本平台旨在为医疗卫生专业人士传递更多医学信息。本平台发布的内容,不能以任何方式取代专业的医疗指导,也不应被视为诊疗建议。如该等信息被用于了解医学信息以外的目的,本平台不承担相关责任。本平台对发布的内容,并不代表同意其描述和观点。若涉及版权问题,烦请权利人与我们联系,我们将尽快处理。

(本网站所有内容,凡注明来源为“医脉通”,版权均归医脉通所有,未经授权,任何媒体、网站或个人不得转载,否则将追究法律责任,授权转载时须注明“来源:医脉通”。本网注明来源为其他媒体的内容为转载,转载仅作观点分享,版权归原作者所有,如有侵犯版权,请及时联系我们。)