近日,全球首个非共价布鲁顿酪氨酸激酶抑制剂(BTKi)匹妥布替尼,正式纳入《国家基本医疗保险、工伤保险和生育保险药品目录》,用于既往接受过至少两种系统性治疗(含BTKi)的复发或难治性套细胞淋巴瘤(R/R MCL)成人患者。这一重要进展,不仅为R/R MCL患者提供了新的治疗选择,也标志着我国的B细胞恶性肿瘤靶向治疗迈入新阶段。故此,医脉通特邀苏州大学附属第一医院吴德沛教授和上海交通大学医学院附属瑞金医院赵维莅教授深度剖析匹妥布替尼的药物作用机制,并围绕BTK通路与抑制剂结构、药学特性与克服耐药的能力、脱靶效应与安全性三大维度进行系统阐述。
BTK是B细胞受体(BCR)信号通路中的关键激酶,在B细胞的发育、成熟、增殖、趋化与粘附等生理过程中发挥核心作用1-2。然而,在慢性淋巴细胞白血病(CLL)/小淋巴细胞淋巴瘤(SLL)、MCL等B细胞恶性肿瘤中,BTK信号通路异常激活,进而抑制肿瘤细胞凋亡,促进其异常增殖1-2。这一核心生物学功能使得BTK成为靶向治疗B细胞恶性肿瘤的理想靶点。
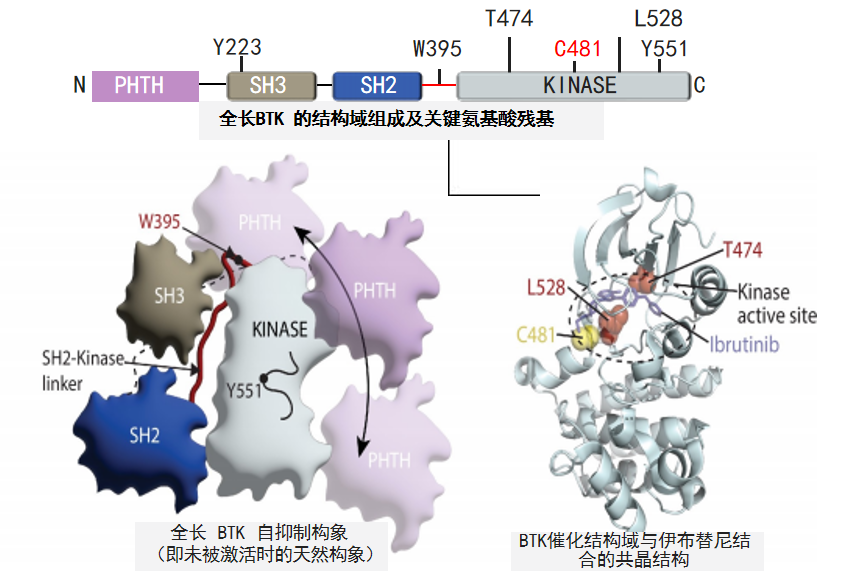
图1 BTKi作用区域及位点
从结构上看,BTK蛋白由659个氨基酸构成,自N端至C端包含PH、TH、SH3、SH2及催化结构域1,3(图1)。催化结构域的ATP结合口袋是BTKi主要作用区,而口袋内的半胱氨酸481残基(C481)是共价BTKi的关键结合位点1,3。目前临床上广泛使用的伊布替尼、阿可替尼、泽布替尼等共价BTKi,均通过不可逆地与C481结合,阻断BTK活性。当C481发生突变时,共价BTKi难以与其有效结合,进而导致耐药。因此,亟需研发新型药物以攻克这一治疗难题。
非共价机制“破局耐药”,优化药代动力学筑牢疗效根基
作为全新一代非共价BTKi,匹妥布替尼在结构与作用机制上实现了对传统共价BTKi的跨越式突破。其创新之处在于不依赖于C481位点的共价结合,而是通过多位点非共价方式与BTK的ATP结合口袋形成广泛的相互作用4,这一独特机制为其卓越的药学特性奠定了结构基础。
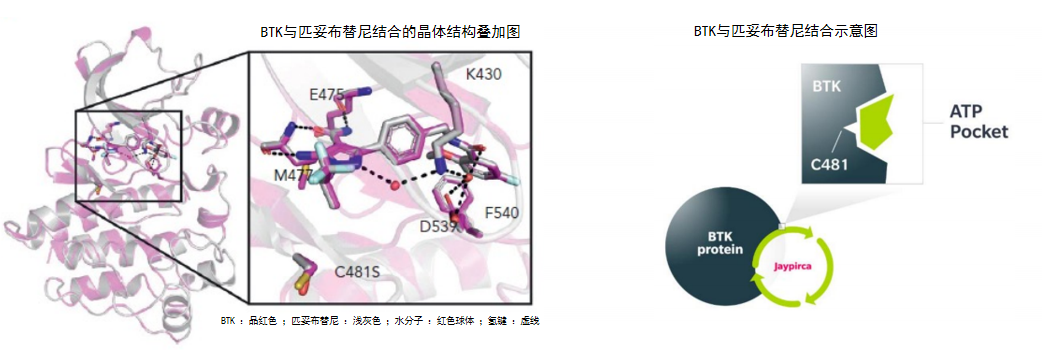
图2 BTK与匹妥布替尼结合的晶体结构叠加图(左)和示意图(右)
从分子层面深入探究,晶体结构研究揭示匹妥布替尼通过3个氢键与ATP结合口袋的E475、M477骨架结合,并与K430、D539形成水分子介导的氢键,进一步与F540形成边对的堆积相互作用4(图2)。值得注意的是,匹妥布替尼与C481的最近距离约为4Å,不存在直接相互作用4,因此C481S突变不会影响其结合模式与抑制效力。更为重要的是,这种非共价结合方式具有柔性调整空间,能够通过动态解离与再结合过程适应BTK局部构象的变化5。另外,氢氘交换质谱分析发现,与共价BTKi不同,匹妥布替尼不仅对BTK激酶结构域的稳定作用更强,还能稳定SH3、SH2结构域紧凑的自抑制构象,使得匹妥布替尼的抑制效果更强3。而结构叠加分析显示,与伊布替尼和泽布替尼相比,匹妥布替尼更深入BTK蛋白,更接近激活环和C螺旋4。并且,匹妥布替尼结合BTK后使BTK稳定在闭合的非活性构象中,这一结构特征使其能够更有效地阻止Y551和Y223等关键位点的磷酸化4,从而有效阻断BTK的激活及下游信号传导。
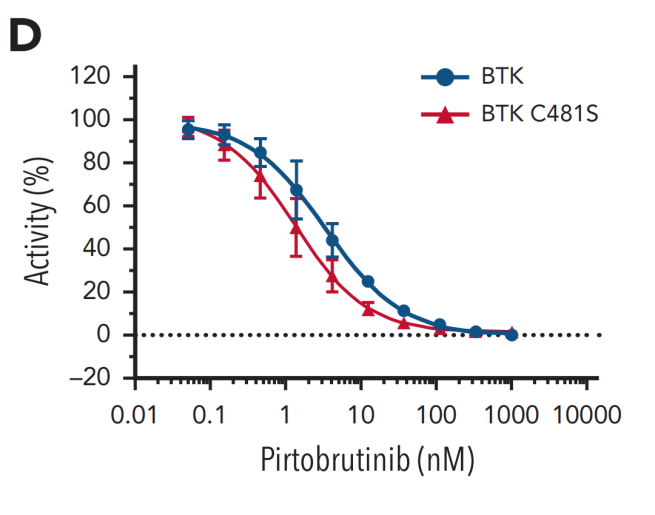
图3 匹妥布替尼对BTK和BTK C481S激酶活性的抑制作用
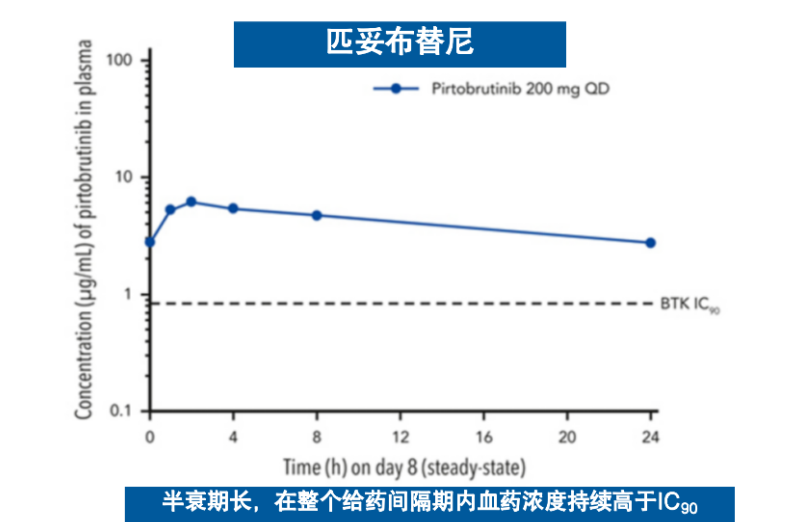
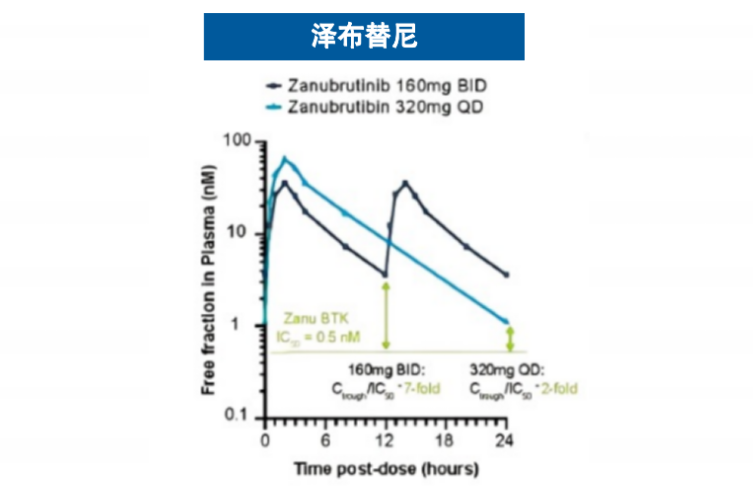
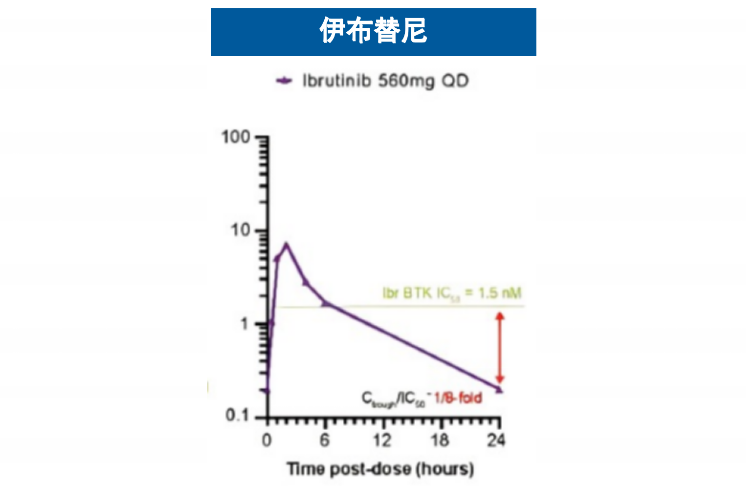
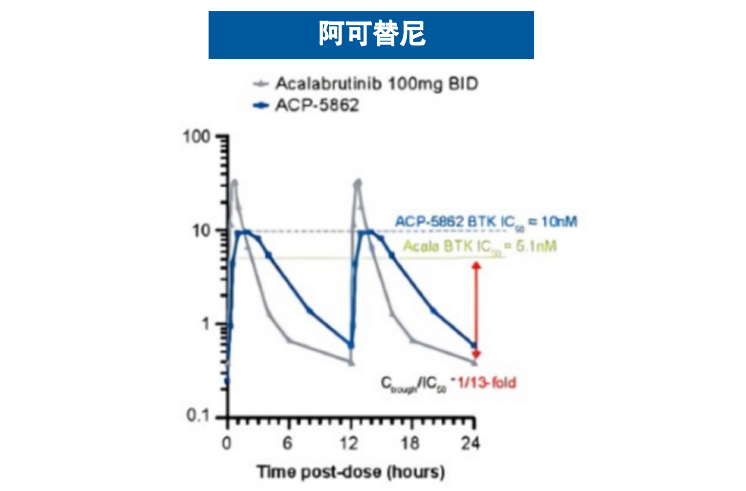
基于这一创新的结合机制,匹妥布替尼展现出卓越的结合特性。其对野生型及C481S突变型BTK均具有个位数纳摩尔级的结合亲和力,且解离速率极低4(图3),这意味着药物与靶点的结合更为稳定、作用时间更为持久。临床前药代动力学数据显示,其半衰期长达19小时,而伊布替尼和泽布替尼的半衰期分别为4-6小时和2-4小时,这一特性支持每日一次给药方案,能够实现24小时持续靶点覆盖6-9(图4),有效避免因药物浓度波动导致的靶点抑制不足现象。
图4 匹妥布替尼和其他共价BTKi的血药浓度曲线
在整体药代动力学特征方面,匹妥布替尼表现出高曲线下面积(AUC)、高绝对生物利用度与低表观分布容积的优良特性。研究数据显示,其AUC高达91300 ng·h/mL,绝对生物利用度为85.5%,而表观分布容积仅为32.8L,显著低于其他BTKi6-8。这一特征提示药物主要分布于血液中,在肝、肾、神经等组织中的暴露程度较低,从而在保持高效抑制BTK活性的同时,显著降低组织特异性毒性的发生风险。
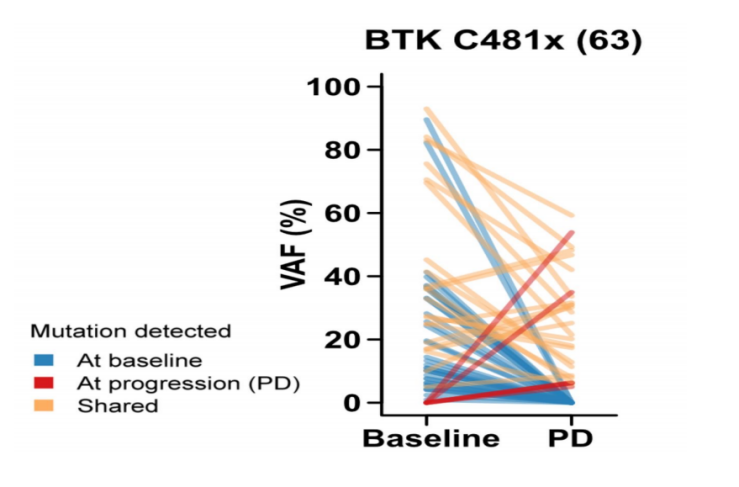
图5 匹妥布替尼清除C481x克隆的能力
这种独特的分子结构和药学优势在克服临床耐药方面展现出显著成效。在表达C481S突变的TMD8细胞系中,匹妥布替尼保持强大的抗增殖活性,IC50为26.4 nM,而共价BTKi在这类细胞中未观察到抗增殖活性4。这一发现得到了临床研究的进一步验证,BRUIN研究结果显示,使用匹妥布替尼治疗后,84%的患者可观察到C481x克隆的减少或清除10(图5),且无论获得的BTK突变类型如何,患者的总缓解率(ORR)均维持在较高水平10,证明了其在多重耐药背景下依然保持的治疗效力。
综合来看,匹妥布替尼通过其非共价结合机制、卓越的药代动力学特性和对突变型BTK的有效抑制,构建了克服临床耐药的多重屏障,为经治B细胞恶性肿瘤患者提供了新的治疗希望。
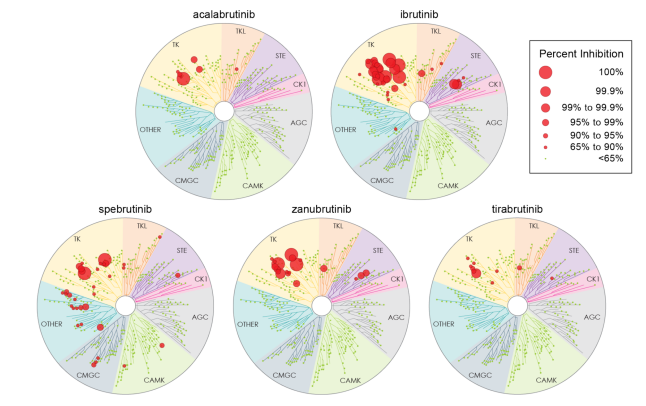
在克服耐药机制的同时,药物的安全性同样是决定其临床应用价值的关键因素。共价BTKi由于结构设计的局限性,往往伴随着显著的脱靶效应,例如对ERBB2/HER2、CSK、EGFR、TEC的抑制可能导致房颤、皮疹、腹泻、出血等不良反应的发生,严重影响了患者的治疗依从性和生活质量(图6)11。
图6 不同BTKi激酶选择性的差异
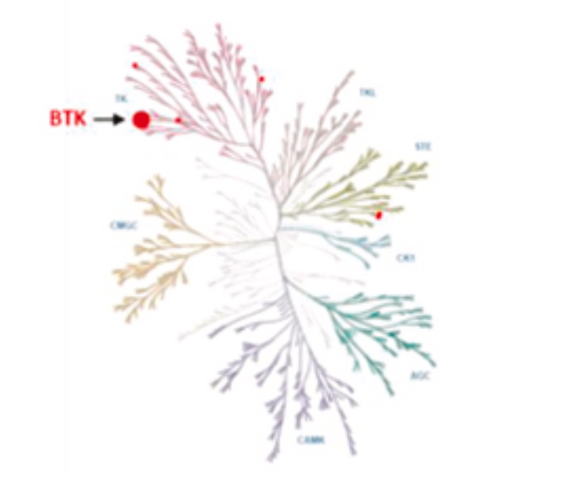
相比之下,匹妥布替尼展现出卓越的靶点选择性(图7,图8)。激酶活性试验显示,在100 nM浓度下,伊布替尼可抑制22种激酶,而匹妥布替尼仅抑制4种激酶,且对BTK的抑制率高达97%4。这种高选择性源于其独特的分子结构设计,使其几乎不与EGFR、TEC、HER2等激酶结合。
图7 匹妥布替尼对BTK激酶的高选择性
临床研究数据进一步证实了匹妥布替尼的安全性优势。匹妥布替尼任何级别的房扑/房颤发生率仅为2.7%,显著降低了心血管不良事件风险6。在肝功能安全性上,伊布替尼在轻、中、重度肝损伤患者中的AUC分别升高2.7倍、8.2倍和9.8倍7,存在明显的药物暴露过量风险。而匹妥布替尼在同类患者中不仅未出现AUC升高,反而在重度肝损伤患者中观察到AUC降低21%的现象6,这一特征使其在肝功能不全患者中具有更好的安全性。
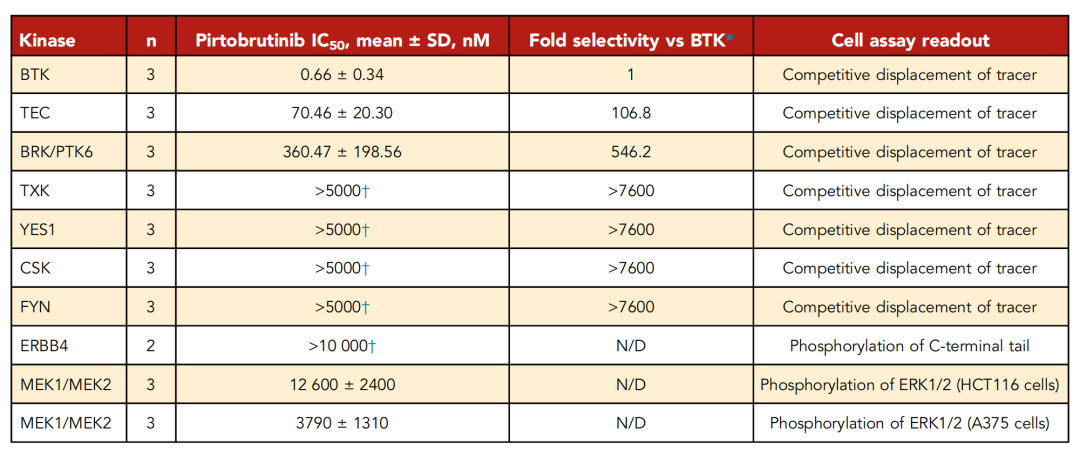
图8 匹妥布替尼在细胞检测中对特定激酶的选择性
另一个值得关注的特性是匹妥布替尼的血脑屏障穿透能力。作为分子量为479.44 Da的脂溶性小分子,其Log P值约为3,血浆蛋白结合率为96%,这些理化特性使其具备良好的中枢神经系统渗透能力6,12。一项针对cBTKi治疗后进展的Bing-Neel综合征患者的临床观察显示,换用匹妥布替尼后,患者可在短期内出现神经症状改善及影像学缓解,提示该药物能有效透过受损的血脑屏障并在中枢神经系统病灶中达到治疗浓度,为其在原发性中枢神经系统淋巴瘤(PCNSL)和治疗后脑转移的临床应用提供了可能性13。
综合来看,匹妥布替尼通过其高度选择性的作用机制和优异的药代动力学特征,实现了从"多靶点抑制"到"精准治疗"的重要转变,为经治B细胞恶性肿瘤患者提供了更为安全有效的治疗选择。
BTKi的迭代发展是精准肿瘤学理念的生动实践。匹妥布替尼作为新一代非共价BTKi,对多种突变型BTK均展现出高效抑制作用。同时,该药物优异的安全性特征进一步提升了其在临床应用中的价值。此次纳入医保目录大幅降低了患者经济负担,使更多复发难治性淋巴瘤患者能够受益于这一创新药物。作为临床医生,我们应当根据患者具体情况进行个体化治疗决策,同时积极积累真实世界用药经验,持续优化治疗策略,共同提升我国B细胞恶性肿瘤诊疗水平,为"健康中国2030"战略目标的实现贡献力量。
匹妥布替尼的上市与医保纳入,是我国淋巴瘤治疗领域的重要进展。其在结构设计上跳出了传统共价结合的框架,通过非共价机制有效克服C481突变导致的耐药问题,这在临床实践中具有重要意义。我们观察到,在既往接受过多线治疗、甚至多种BTKi治疗后进展的患者中,匹妥布替尼仍能带来显著且持久的缓解。其高选择性带来的安全性优势,尤其适合老年、合并心血管疾病或肝功能不全的患者。未来,随着其在更多B细胞肿瘤适应症中的探索,匹妥布替尼有望成为重要支柱药物。
吴德沛 教授
主任医师、教授、博士生导师
苏州大学附属第一医院血液科主任
国家血液系统疾病临床医学研究中心常务副主任
江苏省血液研究所副所长
苏州大学造血干细胞移植研究所所长
首届百名国家杰出医师
全国先进工作者
第十三届、十四届全国政协委员
中华医学会血液学分会第十一届主任委员
中国医药教育协会真菌病专业委员会主任委员
中国造血干细胞捐献者资料库专家委员会副主任委员
参考文献:
1. Wen T, et al. Leukemia. 2021 Feb;35(2):312-332.
2. 陈潇,龚国清. 药学研究,2020,39(03):169-175.
3. Joseph RE, et al. Elife. 2024 Dec 27;13:RP95488.
4. Gomez EB, et al. Blood. 2023 Jul 6;142(1):62-72.
5. RCSB Protein Data Bank. Crystal structure of BTK kinase domain in complex with pirtobrutinib. Retrieved from https://www.rcsb.org/3d-view/8FLL/1
6. 匹妥布替尼产品说明书
7. 伊布替尼产品说明书
8. 泽布替尼产品说明书
9. Thompson P A, et al. Blood, 2023, 141(26): 3137–3142.
10. Brown J R, et al. Blood, 2025: blood.2024027009.
11. Kaptein A, et al. Blood, 2018, 132(Supplement 1): 1871.
12. https://go.drugbank.com/drugs/DB17472
13. Frustaci AM, et al. Am J Hematol. 2025 Oct;100(10):1878-1881.

 我要投稿
我要投稿



















{kind=link}