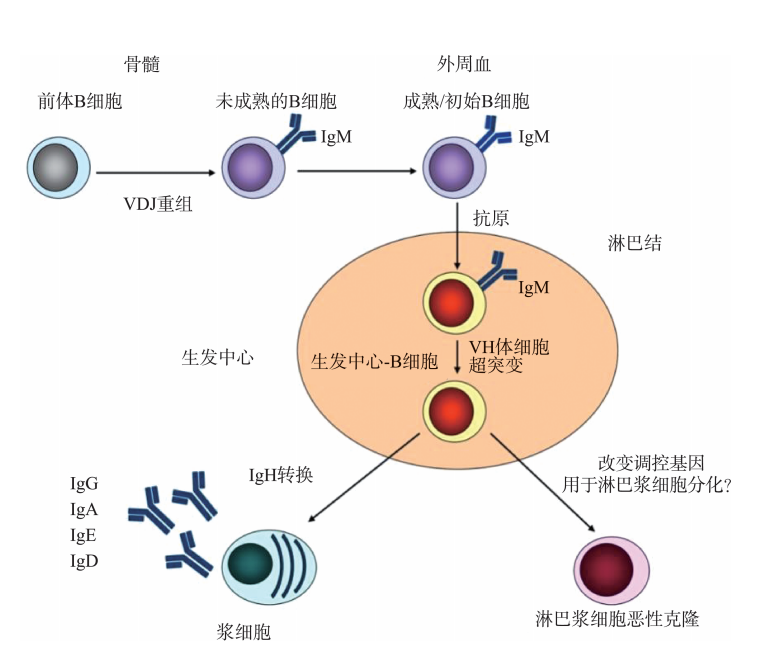
巨球蛋白来自淋巴浆细胞。浆细胞是从B淋巴细胞分化而来,由于基因的调节变化,B细胞可转向淋巴浆细胞方向分化,恶性克隆性淋巴浆细胞导致巨球蛋白血症(图45-1)。
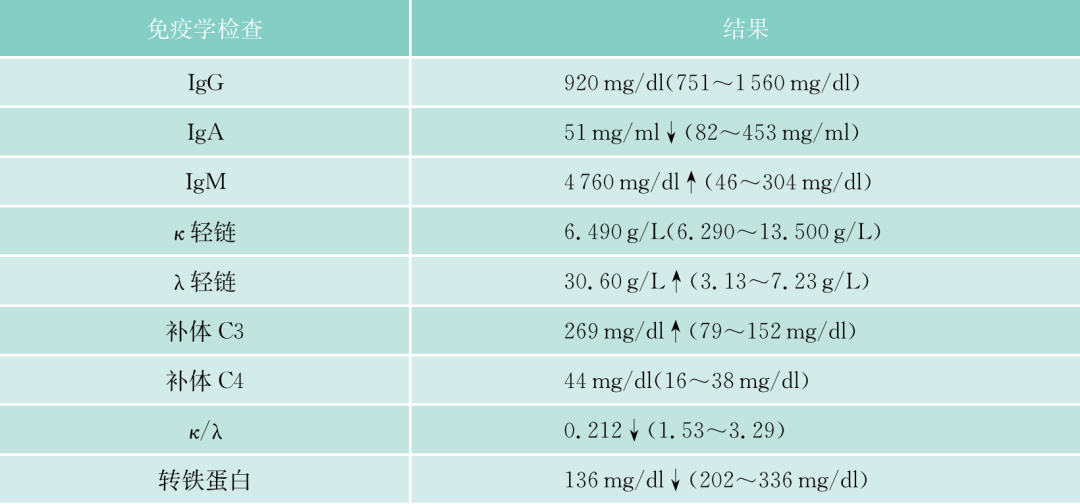
引起巨球蛋白增多的原因很多,病毒感染早期可伴巨球蛋白(IgM)增多,淋巴瘤中少数可伴血巨球蛋白增多(见下文)。克隆性巨球蛋白血症多数为恶性WM,骨髓中所伴随的瘤细胞,多数属浆细胞样淋巴细胞。本病例IgM明显增高,达4760mg/L,血入链增高(30.6mg/L),血清蛋白电泳有M蛋白,尿中入轻链阳性。骨髓中淋巴细胞增多,占43.5%,部分淋巴细胞呈浆阳分化,有的呈浆细胞形态,淋巴细胞免疫表型CD19、CD20、CD22和CD23(+),CD138(一),基因IgH重排阳性。
据上分析,本病例符合WM的诊断标准(表45-7)。
图45-1 前体B细胞分化为浆细胞及淋巴浆细胞过程
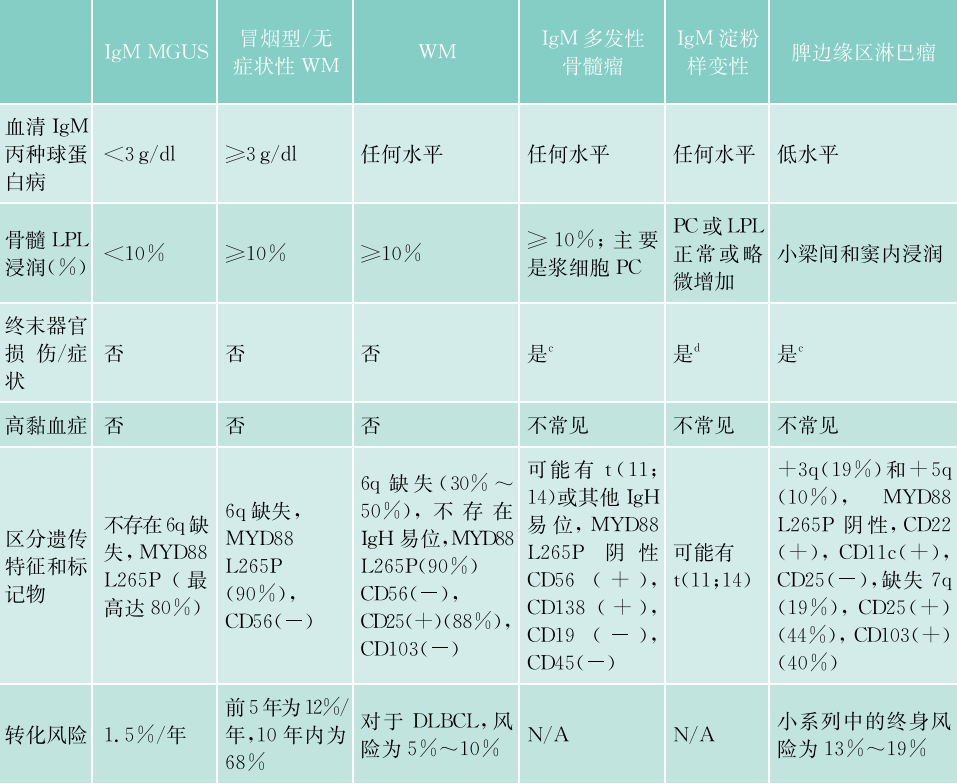
表45-7 WM及相关疾病的诊断标准
MGUS,意义未明单克隆免疫球蛋白血症。
根据发展过程及伴随的病变,克隆性浆细胞样淋巴细胞伴巨球蛋白增高可分为3种:①意义未明单克隆免疫球蛋白血症(MGUS);②冒烟型IgM血症;③巨球蛋白血症或WM。本病例血中巨球蛋白明显增高(达4760mg/L),骨髓中淋巴细胞43%,部分呈浆细胞样变,故已发展为WM。WM又称LPL/WM。
本病必须与伴有IgM的淋巴瘤(如脾边缘区淋巴瘤、套细胞淋巴瘤、滤泡性淋巴瘤)、淀粉样变、冷凝集素性溶血性贫血及IgM型多发性骨髓瘤作鉴别(表45-8),这些淋巴瘤骨髓中可见异型幼淋细胞,淋巴结、脾肿大,血中巨球蛋白可高,但一般低于3g/L。本患者无淋巴结肿大,主要变化是血IgM明显增高(达4760mg/L),伴有λ轻链克隆性增多,无溶血。根据国内诊断多发性骨髓瘤的标准,必须符合:①骨髓中浆细胞>15%并有原浆或幼浆细胞,或组织活检证实为浆细胞瘤。②血清单克隆免疫球蛋白(M蛋白)IgG>15g/L。③广泛骨质疏松和(或)溶骨病变。本病例无溶骨改变,骨髓中出现的细胞是浆样变的淋巴细胞,故不符合IgM型MM。
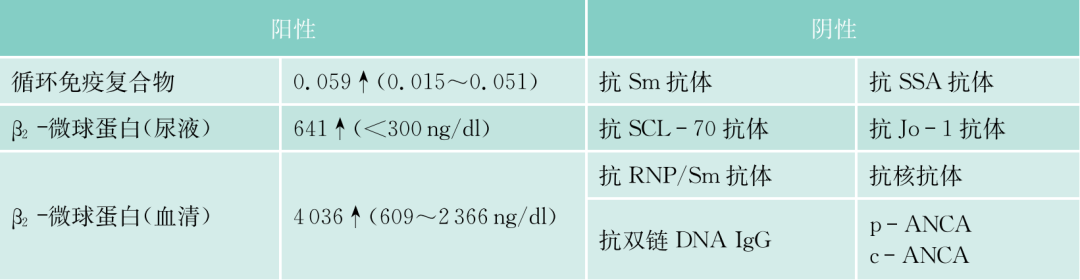
需要排除的其他IgM增多症,尚有IgM型MGUS,IgM-相关的冷凝集素综合征和2型冷球蛋白血症,因无相应的临床表现,可以排除。
表45-8 巨球蛋白血症的各种表现
IgM,免疫球蛋白M;LPL,淋巴浆细胞淋巴瘤;MGUS,意义未明单克隆免疫球蛋白血症;N/A,不适用;PC,浆细胞;WM,华氏巨球蛋白血症;a.表中列出了IgM单克隆丙种球蛋白病的一些重要鉴别诊断。IgM副蛋白几乎可以存在于所有B细胞淋巴组织增生性疾病中;b.全身症状:肝脾肿大、淋巴结病、贫血、高黏血症、实体器官受累和罕见的溶解性病变;c.CRAB特征(高钙血症、肾功能衰竭、贫血和骨病变);d.通常累及的器官是肾脏、心脏、神经、舌、胃肠道和肝脏。IgM淀粉样蛋白轻链(AL)淀粉样变性患者的肺、淋巴结、周围神经受累频率较高,心脏受累频率较低,与非IgM AL淀粉样变性相比,游离轻链的浓度往往较低;e.主要累及脾脏,淋巴结病很罕见
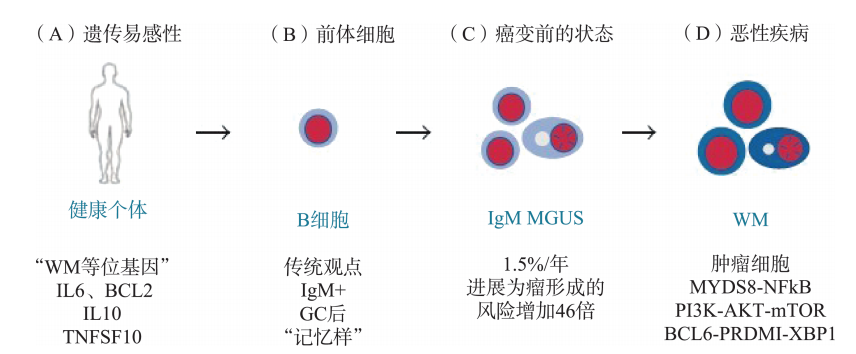
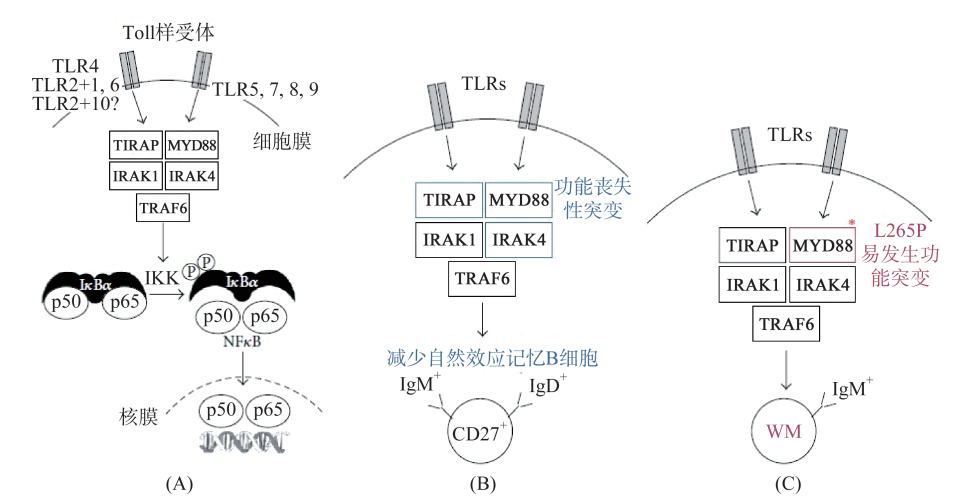
基础研究发现,WM细胞多起源于经过抗原刺激的、源自生发中心的记忆性B细胞,这群细胞分泌IgM。通过遗传易感性基因、一些细胞因子如IL6、IL10的异常参与,经过IgM MGUS阶段,最后形成WM(图45-1、图45-2)。进一步研究发现,基因MYD88的突变决定了疾病的发生。如果MYD88基因发生失功能性的突变,则记忆性B细胞功能将下降;即发生MYD88L265P突变后,将会与IRAK4结合,激活IRAK1以及TRAF6(图45-3),后者进一步活化NFₖB通路,导致疾病发生。研究发现MYD88阳性患者的预后更差。根据最近的研究,90%的WM/幼淋细胞淋巴瘤有MYD88L265P突变,而慢淋只有3%阳性。此外CXCR4 WHIM-like突变占30%~35%。因此,检测MYDL265P基因突变有助于进一步明确WM的诊断。
图45-2 WM的发病过程
图45-3 WM基因突变与发病机制
表45-9 各种指数分期法与预后
根据WM国际预后指数(ISSWM)分期,本患者血红蛋白下降伴有β₂-微球蛋白升高,积分为2分,属中危组,5年生存期为68%。按Mayo Clinic预后分期,因有肝脾肿大,但年龄小于60岁,故10年OS只有16%。
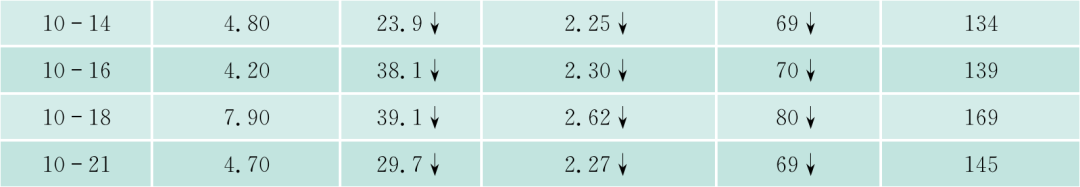
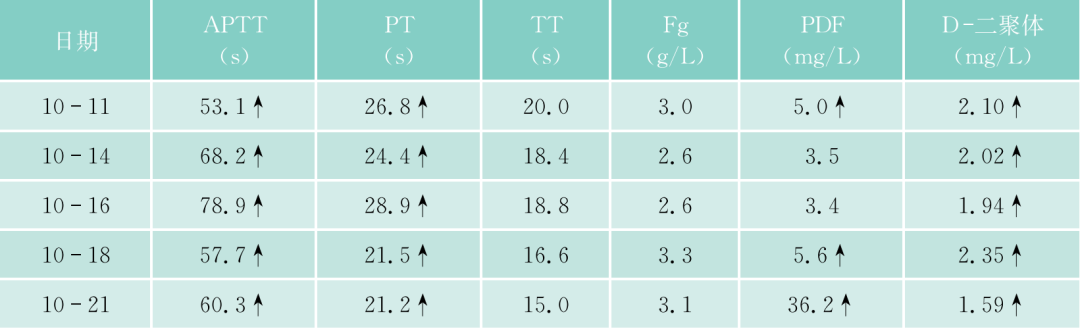
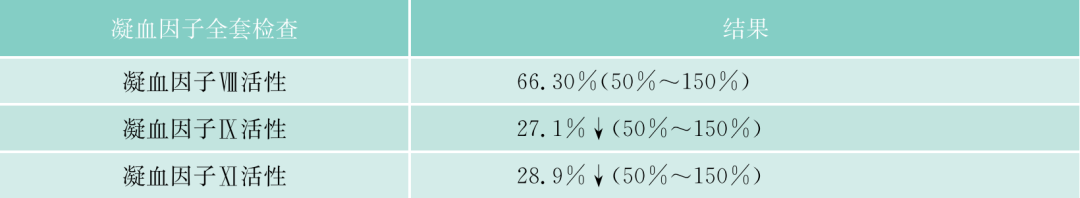
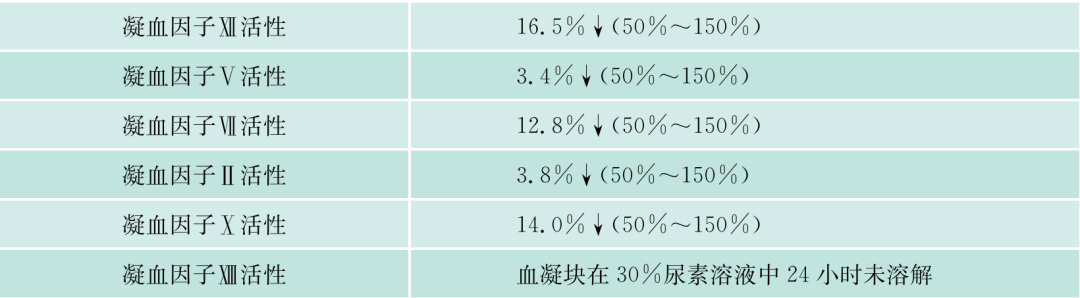
该患者入院后常规检查,发现APTT、PT均明显延长,TT正常,但无出血症状。进一步检验,凝血因子II、V、VII、IX、X、VII、XI、XII都减少,但都不低于正常的2%,FDP一次明显升高达36.2mg/L,但纤维蛋白原不明显降低,D-二聚体升高,但不明显,说明没有DIC,但有纤溶增高,能被代偿。WM本身会否引起凝血因子减少?文献报道,WM可引起免疫性血小板减少、获得性vW综合征。文献上早已报道,实验室检查发现副蛋白血症、伴有获得性凝血异常的发生率者并不少见,原发性淀粉样变、MM、WM及MGUS患者可合并止血功能异常,临床上可无出血症状(亚临床型)。本病例临床上并无出血症状,但实验室检查凝血因子减少,有的十分明显,但均大于正常的2%。什么是凝血因子减少的机制?除了肝的产生减少外(该患者的肝肿大),该患者是否还有淀粉样变?因患者血和尿中轻链都增高,淀粉样变可吸附凝血因子,使这些凝血因子易在肝内破坏。该患者无明显淀粉样变的症状(如肾功能低下,舌大等),故可能性不大,必要时,做肝、皮肤、骨髓活检,行Congo红染色。
随着新药的发现,尤其是靶向治疗药物的应用,WM的治疗有了很快的进展,完全缓解率明显提高(表45-10)。目前BTK抑制剂已被FDA批准用于治疗WM,机制为抑制NFₖB通路的活化,而国内也在积极做临床试验加以验证。根据目前资料,多数患者可由利妥昔单抗联合化疗取得满意疗效。而R-CHOP、R-福达拉滨以及R-万珂-Dx方案效果尤为明显,完全缓解率可超过90%。值得注意的是,单药使用利妥昔单抗短时间内可以出现IgM上升,导致高凝滞血症,故不建议使用单药治疗。建议该患者给予血浆置换后.联合R-CHOP方案治疗,而在CR后给予利妥昔单抗维持治疗。
表45-10 各种治疗WM方案的效果

 我要投稿
我要投稿


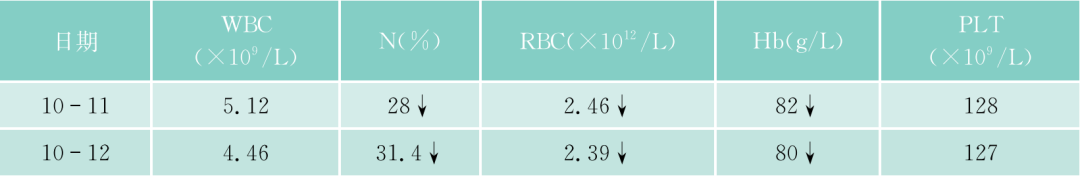












{kind=link}