近日,《美国肾脏病杂志》(AJKD)发布了一例由梅奥诊所(Mayo Clinic)发现的罕见病例:获得性LCAT缺乏症伴IgG3κ单克隆
78岁白人男性患者,因长期进行性
患者为收养儿,无家族病史可查。
未见
抗磷脂酶A2受体(PLA2R)抗体、乙型及丙型肝炎病毒、HIV、抗核抗体检测均为阴性。SPEP/UPEP联合免疫固定电泳未检出单克隆蛋白,sFLC比值正常(1.45,参考范围0.26-1.65)。
第三次骨髓活检未见异常。
外周
肾脏活检显示:单克隆膜性肾病(IgG3κ型),以及符合LCAT缺乏症特征的脂质沉积(图1)。
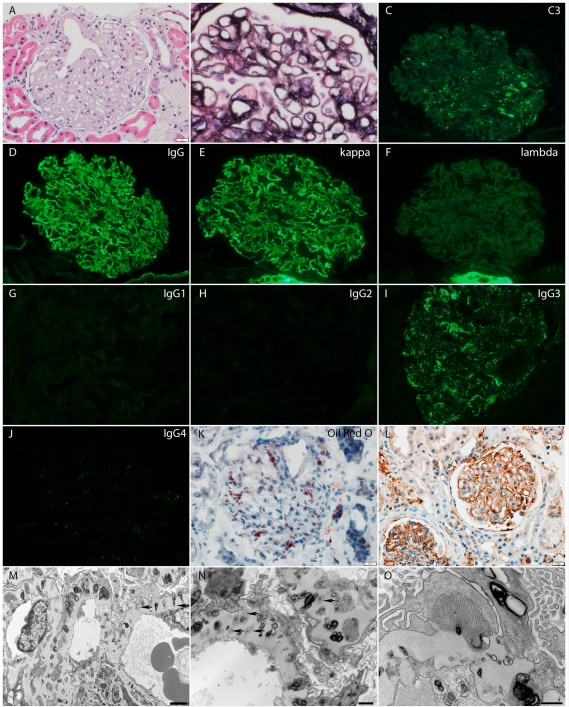
图1 肾脏活检结果
(A、B)光镜显示非增生性肾小球,伴有节段性系膜区增宽及肾小球基底膜增厚(A:苏木精-伊红染色;B:银染色)。银染色可见肾小球基底膜沿线呈全局性空泡化及孔洞形成,系膜区呈节段性空泡化(B)。未见新月体、坏死、节段性硬化或肾小球基底膜双轨征。存在中度肾小管萎缩及间质纤维化,伴极轻度慢性炎症和重度动脉硬化,其中一支动脉可见胆固醇栓子(未展示)。
(C-F)免疫荧光检测显示IgG(D)与κ轻链(E)沿毛细血管壁呈3+弥漫性全局颗粒样着色,C3呈弱阳性节段性着色(C),λ轻链(F)及PLA2R(未展示)染色阴性。
(G-J)IgG亚型免疫荧光染色显示IgG3沿毛细血管壁呈全局颗粒样着色(I),而IgG1(G)、IgG2(H)和IgG4(J)染色均为阴性。
(K)油红O染色突出显示系膜区及沿肾小球基底膜分布的脂滴。
(L)血清淀粉样蛋白P免疫组化染色显示肾小球基底膜呈弥漫性全局颗粒样着色。
(M-O)超微结构观察可见大量上皮下及膜内电子致密沉积物(箭头),伴肾小球基底膜"钉突"反应;同时存在大量嗜锇性上皮下/膜内、节段性系膜区及偶见的内皮下沉积,多数呈圆形层状结构(M、N),少数呈松散线状(O)。足细胞及肾小管上皮细胞内可见少量类似嗜锇性层状沉积物(未展示)。足细胞表现为全局性足突融合。
原始放大倍数:A-L为×400倍,M为×4,800倍,N为×9,300倍,O为×23,000倍。
比例尺:M图为2 μm,N图为1 μm,O图为600 nm。
进一步复查患者的血脂谱。结果显示,患者HDL-C在过去10年持续偏低,波动于<3–12 mg/dL(参考范围 40–59 mg/dL),而甘油三酯和总胆固醇水平均正常。胆固醇酯占总胆固醇的比例仅为11%(参考范围 60%–80%),支持LCAT缺乏症的诊断。血清α-半乳糖苷酶活性正常,可排除法布雷病。
对与原发性高甘油三酯血症及相关疾病相关的基因(APOA5、APOC2、APOE、CREB3L3、GPD1、GPIHBP1、LCAT、LIPA、UPC、LMF1、LPL 和 LRP6,包括对 APOE E2/E2 纯合子的靶向检测)的编码区及内含子/外显子边界区域,以及已知存在致病变异的区域,进行了下一代测序(NGS)和/或 Sanger 测序分析。同时采用 NGS 和/或基于聚合酶链反应(PCR)的定量方法,检测上述基因的缺失和重复变异。结果未检测到可报告的致病变异、可能致病变异或意义未明变异,也未检测到 APOE E2/E2 纯合子。
为了明确肾小球上皮下沉积物的蛋白质组成,对肾小球进行了激光显微切割,随后采用液相色谱-串联质谱(LC-MS/MS)分析。结果显示,沉积物中未检测到任何已知的MN抗原,包括 PLA2R、THSD7A、EXT1/EXT2、NELL1、SEMA3B、CNTN1、NCAM1、PCSK6、HTRA1、PCDH7、FAT1 和 NDNF。然而,质谱在两份独立的显微切割样本中均检测到了LCAT(专属谱图计数分别为 14 和 11)和 SAP(专属谱图计数分别为 140 和 132)。在既往使用相同方法和平台检测的 73 例 PLA2R 阴性膜性肾病(包括 41 例已明确靶抗原者)、2 例 PLA2R 相关膜性肾病、2 例 IgG3 型 PGNMID、1 例伴隐匿性 IgGκ 沉积的膜性样肾小球病(MGMID)、1 例单克隆膜性肾病、1 例 C3 肾小球肾炎、2 例 IgA 肾病、1 例糖尿病肾小球硬化、1 例局灶节段性肾小球硬化以及 1 例抗体介导性排斥反应中,均未检测到 LCAT。SAP 免疫组化染色显示肾小球基底膜(GBM)呈弥漫颗粒状阳性(图1L)。
为了明确肾小球中的脂质成分,研究团队参考近期发表的一项迟发型法布雷病研究方法,对肾小球进行了LC-MS/MS脂质组学分析。热图聚类结果显示,肾小球内的磷脂(主要为磷脂酰胆碱和鞘磷脂)含量升高,而神经酰胺含量降低。磷脂酰胆碱是细胞膜的主要组成成分之一。已知LCAT 可将 1 个酰基从磷脂酰胆碱转移至胆固醇,以合成胆固醇酯。在此过程中,LCAT 还会生成溶血磷脂酰胆碱。本研究中观察到多种磷脂酰胆碱的累积,提示该类脂质在LCAT 代谢途径中存在异常。神经酰胺的减少可能反映LCAT 活性下降,因为已有研究表明,神经酰胺有助于提高磷脂酰胆碱对 LCAT 的可利用性。
患者初始接受赖诺普利治疗,但出现肌酐升高至1.99 mg/dL,故换用氯沙坦。随后其血清肌酐降至1.22 mg/dL,蛋白尿也恢复至正常水平(<200 mg)。此后蛋白尿反弹至1.3 g/g,遂启用达雷妥尤单抗治疗。至肾活检后22个月(启动化疗后15个月),患者蛋白尿为682 mg,血清肌酐为1.63 mg/dL,而高密度脂蛋白水平仍低于5 mg/dL。
本文首次报道了获得性LCAT缺乏症伴IgG3κ单克隆膜性肾病的病例,拓展了具有肾脏意义的单克隆丙种球蛋白病(MGRS)的疾病谱。本病例及既往研究均在肾小球中检测到LCAT蛋白,结合患者肾小球内未检出其他已知MN抗原,提示LCAT可能是一种新的MN靶抗原,应考虑将其纳入MN抗原谱。
参考文献:
Lihong Bu, et al.IgG3κ Monoclonal Membranous Nephropathy Associated With Acquired Lecithin Cholesterol Acyltransferase Deficiency.American Journal of Kidney Diseases,Volume 86, Issue 6,2025,Pages 854-859,ISSN 0272-6386.
医脉通是专业的在线医生平台,“感知世界医学脉搏,助力中国临床决策”是平台的使命。医脉通旗下拥有「临床指南」「用药参考」「医学文献王」「医知源」「e研通」「e脉播」等系列产品,全面满足医学工作者临床决策、获取新知及提升科研效率等方面的需求。仅供HCP观看。

 我要投稿
我要投稿















{kind=link}