
“匠心求索,彰显华夏硕果”
ATS现场,董航明教授团队接受采访
Pde4b 抑制剂 Nerandomilast 通过抑制巨噬细胞胞外陷阱释放改善激素抵抗型哮喘


研究背景
本次汇报的研究课题聚焦于激素抵抗型哮喘的发病机制及潜在干预策略。
首先,流行病学数据显示,全球范围内哮喘患者约2.6亿,年死亡人数近50万。其中约5%~10%的患者对高剂量吸入性糖皮质激素反应欠佳,临床定义为激素抵抗型哮喘。该类患者症状持续、反复急性发作,却占据哮喘相关医疗总支出的近50%。其气道炎症特征并非经典的嗜酸粒细胞增高,而是以中性粒细胞浸润为主,目前临床上缺乏有效的治疗手段。文献报道及我们前期的研究发现,巨噬细胞在激素抵抗型哮喘中发挥重要作用。
其中,巨噬细胞胞外陷阱是一种由DNA和弹性蛋白酶等构成的网状结构,可能介导了持续且难以控制的炎症循环。已有证据表明,在重度哮喘患者中,巨噬细胞胞外陷阱释放增加,并通过增强中性粒细胞气道炎症而加重疾病严重程度。基于此,我们提出科学问题:是否存在药物能够靶向这一病理环节?
我们关注到一种口服、高选择性的PDE4B抑制剂——Nerandomilast。该药物最初针对
综合上述两点,我们提出科学假设:若巨噬细胞胞外陷阱是激素抵抗的关键驱动因素,则通过Nerandomilast抑制PDE4B,可能同时减轻中性粒细胞炎症和气道重塑,从而对这一难治性哮喘亚型产生治疗效益。
研究方法
本研究首先收集激素抵抗型哮喘患者临床样本(诱导痰等),通过Western Blot、免疫荧光检等测PDE4B及METs表达。体外使用炎症因子刺激THP-1巨噬细胞,Western blot验证PDE4B蛋白变化。
研究结果
1.PDE4B在激素抵抗型哮喘中表达升高
在激素抵抗型哮喘患者的肺泡灌洗液转录组及诱导痰样本中,均检测到PDE4B表达显著上调。体内实验中,基于自建的LPS联合OVA诱导的激素抵抗型哮喘小鼠模型,均确认巨噬细胞中PDE4B表达升高,且免疫荧光显示PDE4B与巨噬细胞标志物F4/80的共定位增加。
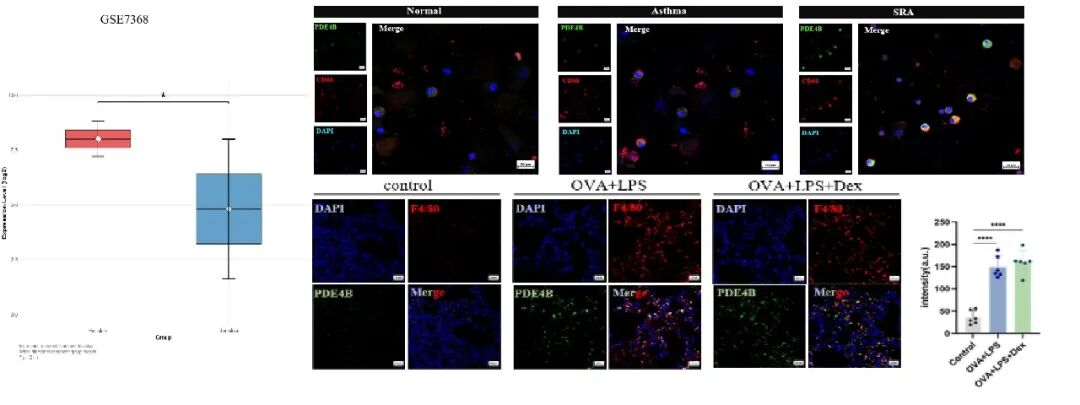
2. Nerandomilast在激素抵抗型哮喘小鼠模型中可抑制炎症与气道重塑
给予Nerandomilast后,能明显抑制二型和非二型炎症因子。肺组织病理学显示,HE与PAS染色证实炎性细胞浸润和粘液分泌减少。Masson染色及胶原蛋白、α‑SMA免疫组化提示气道胶原沉积及重塑明显减轻。
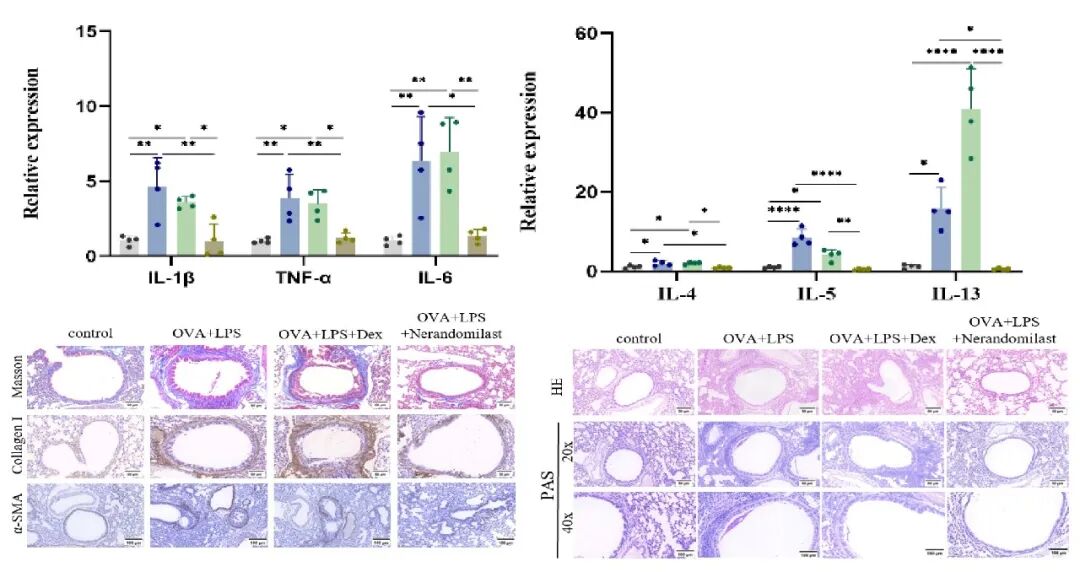
3.巨噬细胞胞外陷阱升高是激素抵抗型哮喘的重要病理特征
在SRA小鼠模型中,肺组织F4/80与citH3共定位及血浆ELISA检测同样证实METs水平升高。给予Nerandomilast后,METs释放显著减少,其效应与已知METs抑制剂CI‑Amidine相当。体外实验中,LPS刺激巨噬细胞后采用SYTOX-Green染色及Western blot均证实Nerandomilast可有效抑制METs的形成。
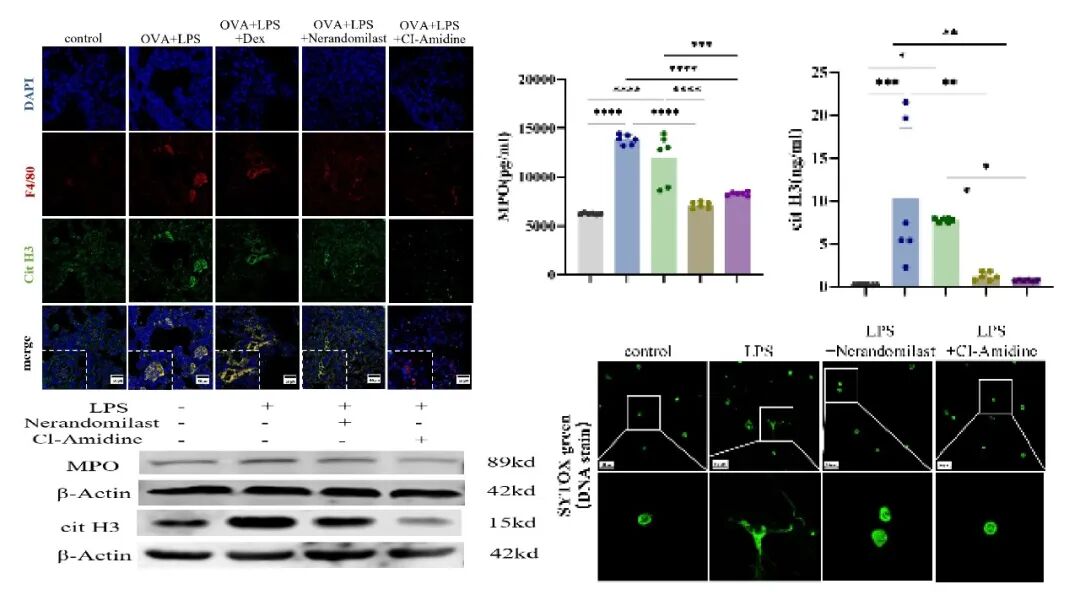
4.抑制巨噬细胞胞外陷阱是Nerandomilast缓解激素抵抗型哮喘的关键机制
单独使用METs抑制剂CI‑Amidine干预后,HE、Masson染色、α‑SMA免疫组化、以及Western blot对气道重塑指标的检测均证实,CI‑Amidine可明显抑制气道重塑,且效应与Nerandomilast高度一致。上述结果反向证实:METs是Nerandomilast在激素抵抗型哮喘中发挥抗炎及抗重塑作用的关键靶点。
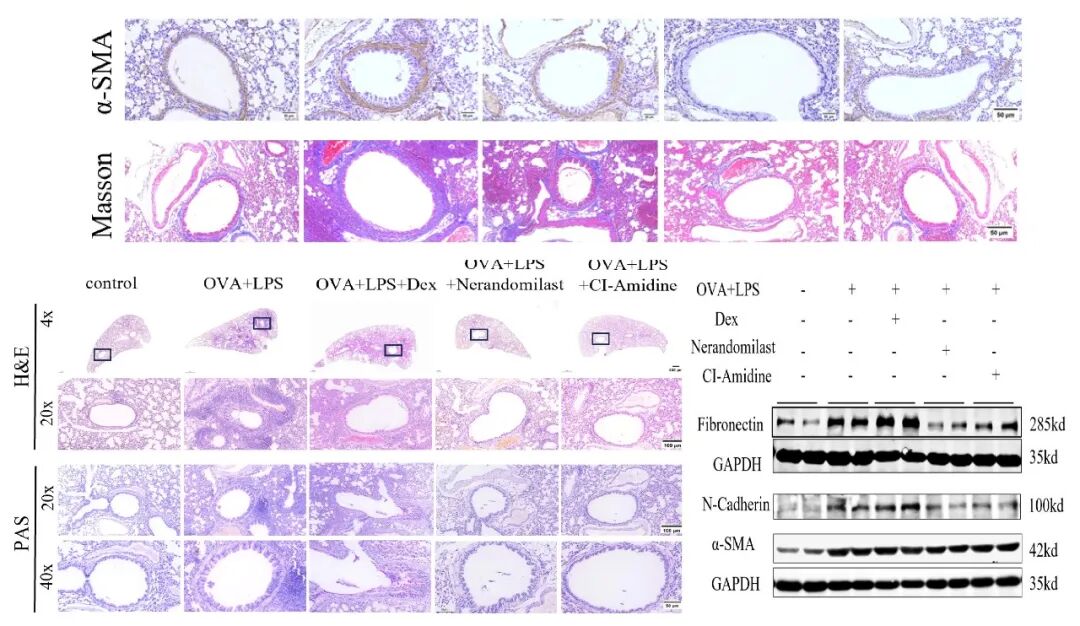
研究结论
在激素抵抗型哮喘中,巨噬细胞通过释放METs驱动中性粒细胞为主的持续性气道炎症及气道重塑。PDE4B抑制剂Nerandomilast可能通过抑制METs的形成,同时实现对炎症和重塑的控制。该研究为激素抵抗型哮喘患者提供了新的治疗理论依据。
RUNX3甲基化同步驱动肺纤维化与肺癌糖酵解及EMT


研究背景
特发性肺纤维化与肺癌常合并发生,二者共享多种危险因素及分子病理特征,提示其可能源于肺泡上皮的共同致病起源。我们假设:抑癌基因RUNX3的表观遗传失活是一个统一的启动事件,同步驱动两种疾病的发生发展。
研究方法
通过MSP、qPCR、Western blot、ChIP-qPCR、双荧光素酶报告基因等实验,分析RUNX3甲基化状态及RUNX3下调后的转录调控。
研究结果
疾病状态下肺泡上皮细胞中DNMT3a表达升高,可直接结合于RUNX3启动子促进其甲基化,导致RUNX3沉默。RUNX3下调后,其下游靶基因GLUT3(SLC2A3)表达上调,进而增强肺泡上皮细胞的糖酵解水平及EMT程序。
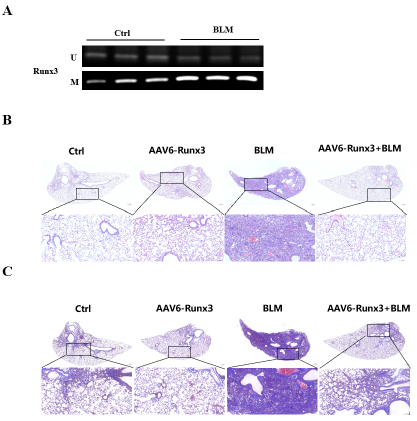
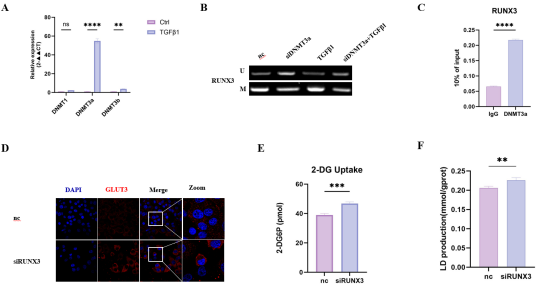
研究结论
DNMT3a介导的RUNX3甲基化通过解除对GLUT3的转录抑制,同步激活糖酵解与EMT,从而协同驱动IPF与LC的发病。RUNX3甲基化有望成为IPF的早期生物标志物,以及两种疾病共有的治疗靶点。
人参来源的纳米粒子可增强


研究背景
哮喘影响全球超过3亿人,目前尚无根治方法。吸入性糖皮质激素如布地奈德(BUD)是临床一线控制药物,但长期使用大剂量会带来严重的全身性副作用——糖代谢紊乱、
研究方法
通过利用人参衍生的纳米颗粒(GDNPs)作为载体,将布地奈德包载其中,构建可吸入的纳米药物平台BUD@GDNPs。人参本身是传统中药活性成分,具有免疫调节和抗炎作用,可作为"协同载体",同时降低药物副作用。建立了屋尘螨(HDM)诱导的过敏性哮喘小鼠模型,通过滴鼻吸入给药,系统评价疗效,并设置长期高剂量给药组重点观察全身毒副作用。
研究结果
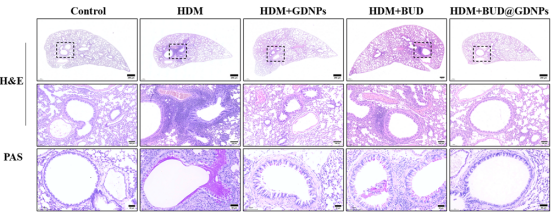
对小鼠肺组织进行组织学染色,结果显示与游离布地奈德相比,BUD@GDNPs显著减轻了气道炎症和黏液分泌。
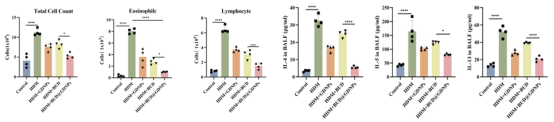
BALF分析证实,BUD@GDNPs组的炎症细胞总数、嗜酸性粒细胞及Th2细胞因子下降更为明显,局部抗炎效果优于游离药物。
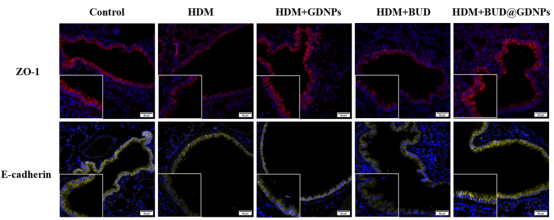
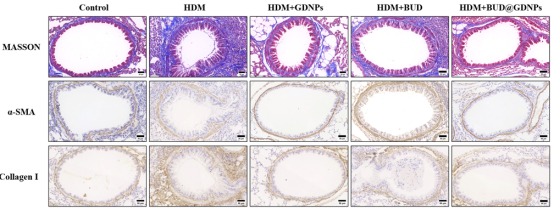
BUD@GDNPs能有效恢复上皮紧密连接蛋白E-cadherin和ZO-1的表达,同时显著降低α-SMA和I型胶原蛋白的沉积。说明该纳米系统不仅能抗炎,还能保护气道结构、缓解气道重塑。
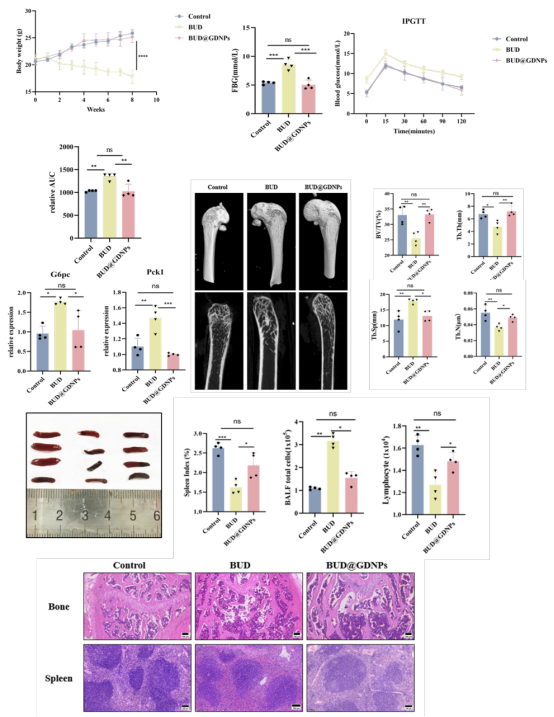
长期高剂量布地奈德导致小鼠出现明显全身毒性:体重下降、血糖升高、肝酶异常、骨小梁丢失、皮质骨变薄、脾脏萎缩、淋巴细胞减少。而BUD@GDNPs几乎完全缓解了所有这些系统性不良反应,各项指标恢复至接近正常水平。这表明纳米递送系统成功做到了"增效"与"减毒"的统一。
研究结论
本研究开发了一种基于人参纳米颗粒的布地奈德吸入递送系统。该系统通过增强局部抗炎效果和减少全身药物暴露,有效控制了气道炎症和重塑,同时显著缓解了糖代谢紊乱、骨质疏松和免疫抑制等副作用。这一双效纳米平台为克服传统吸入性糖皮质激素的关键局限性提供了有前景的新策略。未来我们将进一步优化制剂工艺,并推进临床转化研究。
CAMKK1作为NAT10-ac4C依赖性肺纤维化病理代谢重编程的枢纽


研究背景
特发性肺纤维化是一种严重肺部疾病,其特征为进行性细胞外基质重塑。代谢重编程已逐渐被认为是纤维化进展的重要特征,但连接上游调控信号与下游代谢改变的分子枢纽仍未明确。本研究旨在探索CAMKK1在肺纤维化病理代谢重编程中的作用及其上游调控机制。
研究方法
研究采用整合多组学分析和遗传学验证,并在博来霉素诱导的小鼠肺纤维化模型中开展AAV介导的干预实验,在人肺成纤维细胞中采用siRNA方法进行验证。同时,人IPF标本用于临床相关性分析。
研究结果
CAMKK1在纤维化肺组织中持续上调,并被功能实验证实可驱动疾病发生发展:CAMKK1遗传敲除可减轻纤维化,而CAMKK1过表达则加重病理改变。
机制研究显示,NAT10介导的ac4C修饰是上游表观转录开关,可稳定CAMKK1转录本并增强其翻译效率。结构-功能分析进一步证明,该调控轴依赖完整的NAT10催化活性。
在下游机制方面,CAMKK1通过AMPK介导的SLC7A2转运体激活,协调代谢重编程,从而重塑精氨酸代谢并促进胶原过度生成。
研究结论
该研究将CAMKK1界定为连接NAT10依赖性表观转录信号与肺纤维化病理性代谢重编程的核心处理节点,并提示其可能成为具有临床相关性的治疗干预靶点。
董航明 教授
禹启
南方医科大学南方医院呼吸与危重症医学科博士
王青梅
南方医科大学南方医院呼吸与危重症医学科硕士
杨会敏
南方医科大学南方医院呼吸与危重症医学科博士
吴珠曼
南方医科大学南方医院呼吸与危重症医学科硕士
参考文献
本平台旨在为医疗卫生专业人士传递更多医学信息。本平台发布的内容,不能以任何方式取代专业的医疗指导,也不应被视为诊疗建议。如该等信息被用于了解医学信息以外的目的,本平台不承担相关责任。本平台对发布的内容,并不代表同意其描述和观点。若涉及版权问题,烦请权利人与我们联系,我们将尽快处理。
 我要投稿
我要投稿




















{kind=link}