5月2日至5日,2026年美国消化疾病周(DDW 2026)在美国芝加哥盛大召开。作为全球消化病学领域的“学术风向标”,本届大会以“Discovery at Every Turn”(处处兼有发现)为主题,汇聚领域顶尖专家学者,集中呈现了从基础机制到临床转化的一系列前沿成果,为参会者带来了一场充满启迪的“发现之旅”。
本届大会上,米吉珠单抗、利生奇珠单抗、
要点一览

研究一:米吉珠单抗在IBD中的长期安全性:UC和CD III期研究的整合分析
(Presentation Number: 34)
米吉珠单抗(MIRI)是一种抗白细胞介素-23(IL-23)p19单克隆抗体,已获批用于治疗UC和CD。本研究汇总MIRI在UC和CD中的III期临床试验数据,以更好地了解其长期安全性特征。
汇总中重度活动性UC(N=1371;LUCENT-1/2/3)及CD(N=1033;VIVID-1/2)成人患者数据,最长治疗4年。对UC或CD及整体IBD人群的安全性事件进行汇总统计。安全性结果数据按治疗年份评估,以暴露调整发病率(EAIRs)表示,即每100患者暴露年(PYE)的事件发生率。
在所有试验中,共2404例患者(41.4%女性,中位年龄38.0岁)接受了MIRI治疗,累计5967.9 PYE。UC或CD成人的总体安全性特征相似。在所有IBD患者中,治疗期间出现的不良事件(TEAE)发生率为114.6/100 PYE,从第1年的165.3/100 PYE下降至第4年的79.2/100(表)。观察到的严重不良事件(SAE)发生率在第4年也普遍下降(表)。
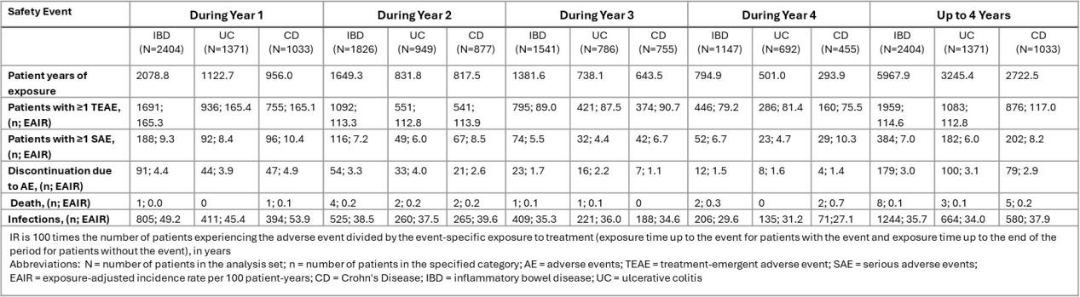
在4年的MIRI治疗期间,严重感染、主要不良心血管事件(MACE)、恶性肿瘤和肝脏事件的发生率持续维持在较低水平,且未随治疗暴露时间延长而增加(图)。
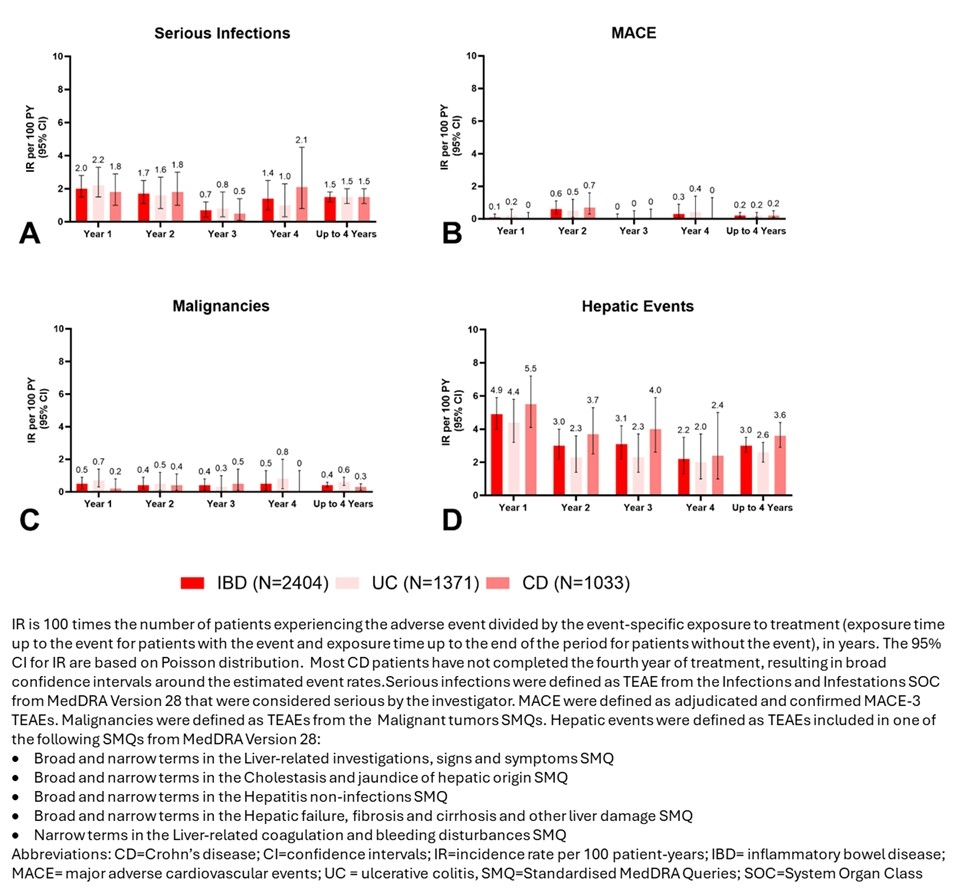
在这项综合分析中未发现新的安全性信号,特别关注的安全性事件发生率在长期用药期间保持稳定。MIRI在整体IBD人群中长达4年治疗的长期安全性稳定,并且在UC和CD适应证中保持一致。
研究二:利生奇珠单抗诱导和维持治疗儿童克罗恩病患者的真实世界疗效和安全性:一项多中心回顾性分析
(Presentation Number: 99)
利生奇珠单抗是一种选择性IL-23 p19抑制剂,已被批准用于成人CD,但儿科证据有限。本研究在多个国际中心评估了利生奇珠单抗在CD儿童中的真实世界有效性、持久性、安全性和给药方案。
该研究是一项回顾性、多中心队列研究,纳入北美、欧洲和中东十个中心在2022-2025年间开始接受利生奇珠单抗单药治疗的<18岁患者。排除同时接受其他先进疗法或既往接受过回盲部切除术或造口术的患者。
主要终点为基线疾病活动(加权儿童克罗恩病活动指数[wPCDAI]≥12.5)患者诱导期后(第12周)达到无皮质类固醇和无完全
次要终点包括临床应答(wPCDAI下降≥17.5)、C反应蛋白(CRP)缓解(<5mg/L)、CFR+CRP缓解、第30/54周的CFR、肠道超声(IUS)缓解、治疗持久性和不良事件(AE)。
共纳入96例儿童(表;中位年龄15岁;51%男性;70%回结肠型;38%既往暴露于乌司奴单抗)。多数(97%)接受成人诱导方案;3例体重<30kg的儿童接受383(IQR 339–397)mg/m²剂量。基线时,71%患儿处于疾病活动期。
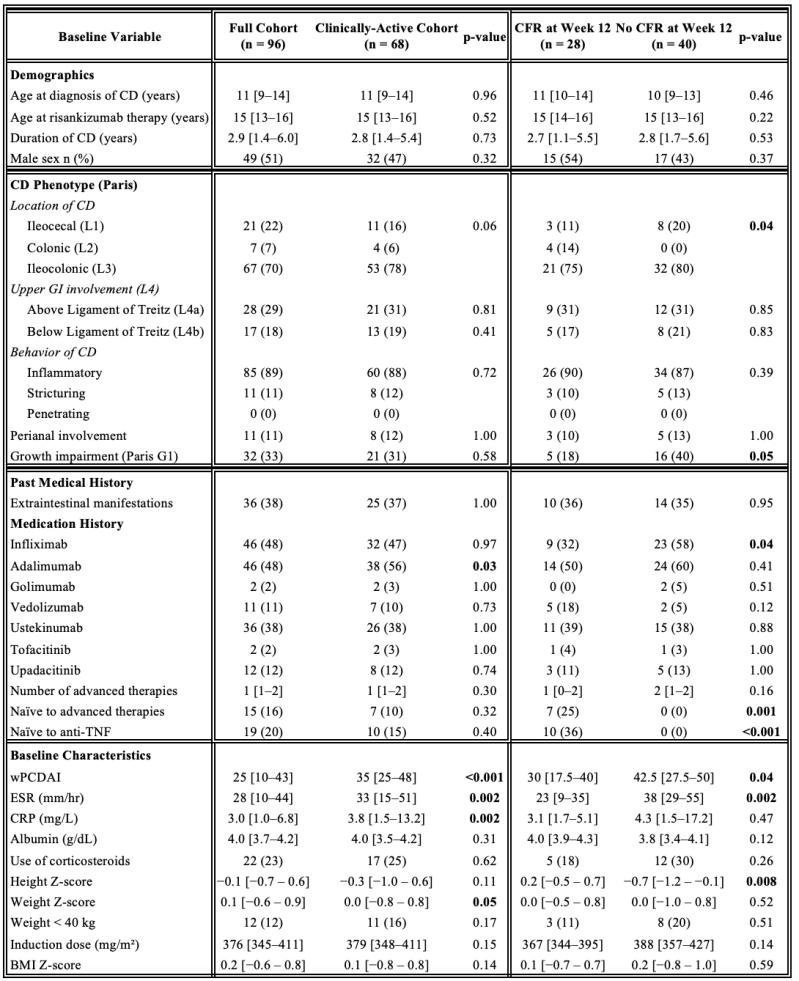
诱导治疗后(疾病活动队列):CFR 41%;临床应答 54%;CRP缓解 66%;CFR+CRP缓解 32%。
维持期(疾病活动队列):第30周CFR 54%,第54周CFR 48%;在诱导后缓解者中,分别有94%和77%在第30周和第54周仍保持缓解。
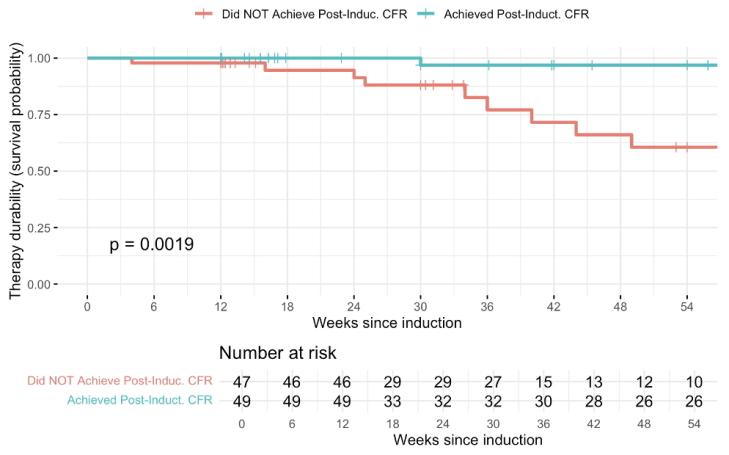
整体队列:中位随访时间33周;88%仍在接受治疗。诱导后达到CFR的患者治疗持久性更高(图;log-rank p=0.002);停药调整后风险比为0.14(95% CI:0.03–0.69;p=0.02)。
较低的基线红细胞沉降率预示较高的CFR几率(OR 0.97 per mm/hr;95% CI:0.94–0.99;p=0.01;Youden≈28.5)。基线IUS活动的患者中,40%达到IUS缓解,与CFR一致(p=0.03)。未报告严重感染、恶性肿瘤或血栓栓塞事件。
在这项首个全球性儿童队列中,利生奇珠单抗实现了有意义的诱导和维持期疗效,具有较高的治疗持久性和良好的安全性。基线炎症负荷影响早期应答,而达到诱导后CFR强烈预测治疗持续性。研究结果支持利生奇珠单抗作为儿童中重度CD的一种安全有效的治疗选择。
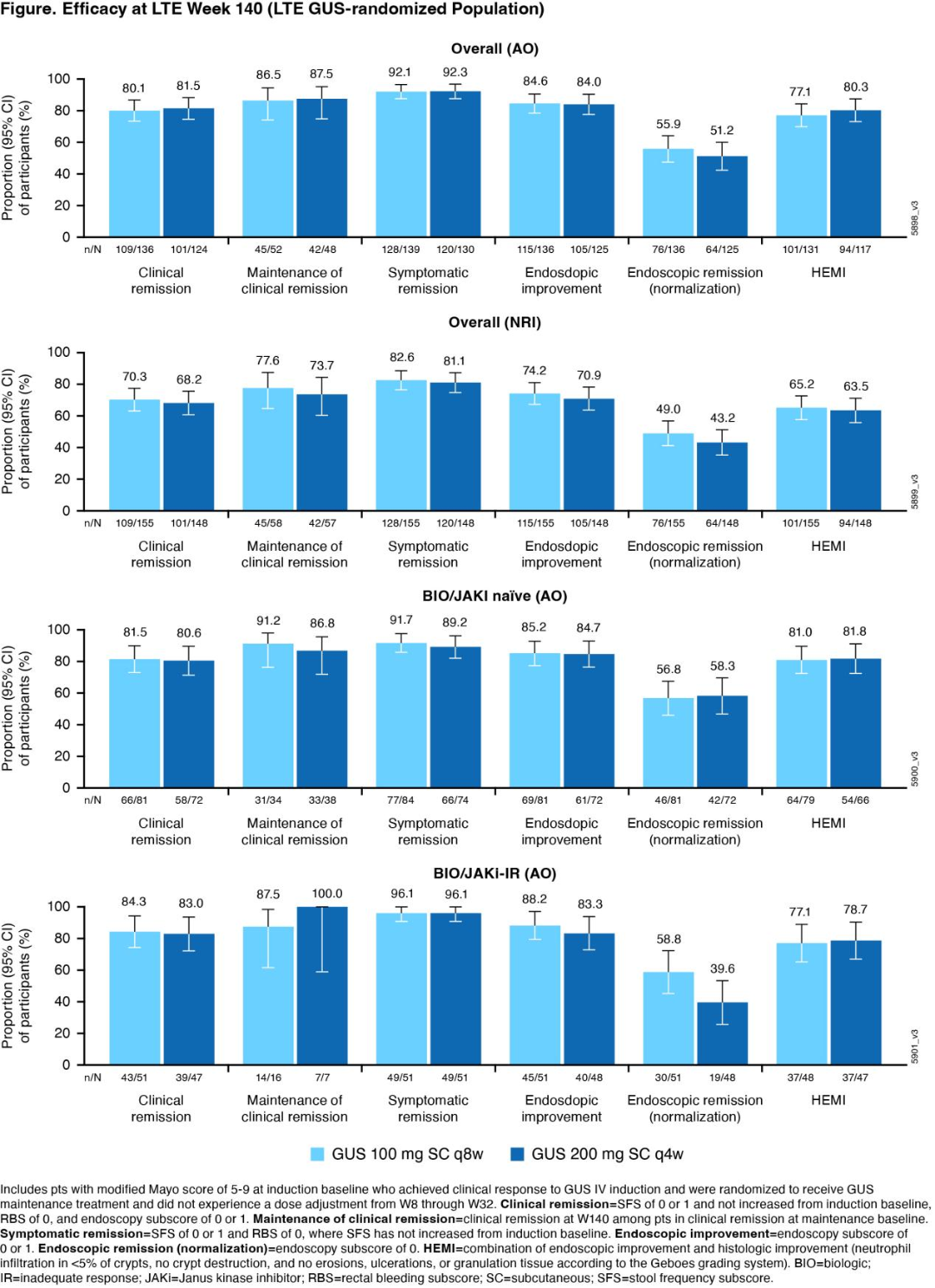
研究三:古塞奇尤单抗治疗溃疡性结肠炎的疗效和安全性:QUASAR长期扩展研究140周数据
(Presentation Number: Su1645)
QUASAR是一项评估古塞奇尤单抗(GUS)治疗中重度活动性UC患者的2b/3期项目。GUS是一种选择性双重作用IL-23p19亚基抑制剂,可有效中和IL-23并与CD64(产生IL-23的细胞上受体)结合。本文报道QUASAR长期拓展研究(LTE)随访至140周的疗效与安全性数据。
对GUS静脉注射(IV)诱导治疗应答者被随机分配接受皮下(SC)维持治疗:GUS 100mg每8周一次(GUS 100);GUS 200mg每4周一次(GUS 200);或安慰剂(PBO)。患者可在第8-32周期间调整剂量至GUS 200。完成44周随访的患者可进入LTE,继续原有治疗方案。研究揭盲后PBO组患者停止治疗。
研究评估了随机分配至GUS组且进入LTE时未进行剂量调整的患者疗效。数据分析方法包括观察法(AO)和无应答者填补(NRI),纳入治疗失败或数据缺失的患者。安全性在所有接受LTE治疗的患者中进行评估。
87.0%的GUS随机化患者进入了LTE,其中88.8%完成了140周治疗。在140周时,GUS 100和GUS 200组患者分别有80.1%和81.5%达到临床缓解,基线(W0)临床缓解者中分别有86.5%和87.5%在第140周维持临床缓解(图;AO)。
在第140周达到临床缓解的210例患者中,205例(97.6%)在第140周前≥8周未使用皮质类固醇。在第140周时,GUS 100和GUS 200组患者中,分别有92.1%和92.3%达到症状缓解,84.6%和84.0%达到内镜改善,55.9%和51.2%达到内镜缓解,77.1%和80.3%达到组织学-内镜黏膜改善(图;AO)。
NRI结果数值上低于AO结果。无论既往是否接受过生物制剂和/或JAK抑制剂治疗,疗效均一致(图)。
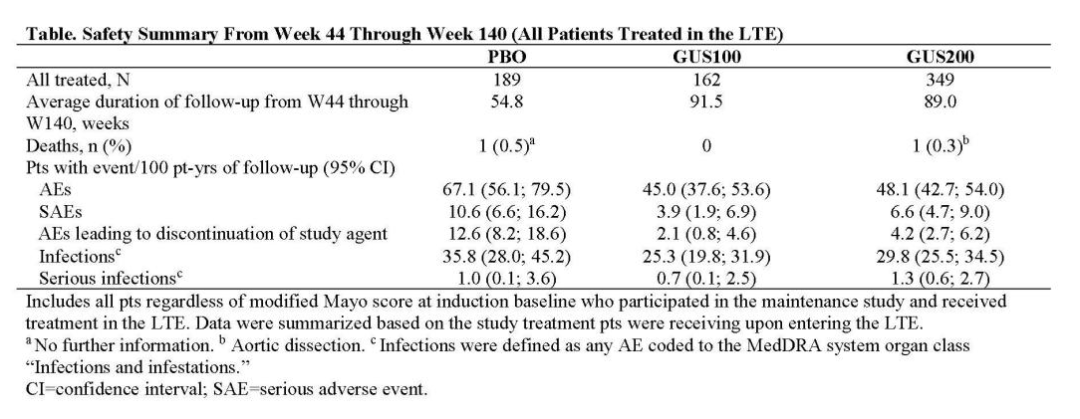
每100患者年中发生≥1次不良事件(AE)的患者人数在GUS组相似(GUS 100组45.0;GUS 200组48.1),而PBO组数值更高(67.1)(表)。GUS治疗患者中报告了1例死亡(
两种GUS维持剂量方案在UC患者中均显示出持续至第140周的临床、内镜和组织学疗效。无论既往是否接受过生物制剂和/或JAK抑制剂治疗,疗效均持续存在。尽管NRI结果数值上低于AO结果,但由于维持期和LTE期间的高保留率,总体趋势一致。未观察到新的安全性问题。
研究四:静脉输注维得利珠单抗在中重度溃疡性结肠炎儿童患者中的疗效与安全性:KEPLER 3期试验结果
(Presentation Number: Su1627)
针对儿童和青少年UC的获批治疗方案有限。KEPLER试验评估了维得利珠单抗(VDZ;一种抗α4β7整合素生物制剂)作为静脉注射(IV)诱导和维持疗法,在儿童和青少年中重度UC患者中的疗效、免疫原性、药代动力学(PK)和安全性。
本研究为3期、单臂试验,纳入2-17岁、体重≥10kg的中重度UC患者(改良Mayo评分5-9分且内镜评分≥2),且既往接受类固醇、免疫调节剂和/或肿瘤坏死因子(TNF)拮抗剂等常规治疗失败。
患者在为期14周的开放标签诱导期接受VDZ IV治疗,随后进入为期40周的随机、双盲维持期,按体重(10-15kg [100 vs 150mg];>15-<30kg [100 vs 200mg];≥30kg [150 vs 300mg])1:1随机分配接受低剂量(LD)或高剂量(HD)VDZ,每8周给药一次。
主要终点是意向性治疗维持人群中第54周时达到改良Mayo评分评估的临床缓解。次要终点包括第14周的临床缓解、持续临床缓解(第14周和第54周)、第54周的无皮质类固醇临床缓解、免疫原性、PK和安全性。
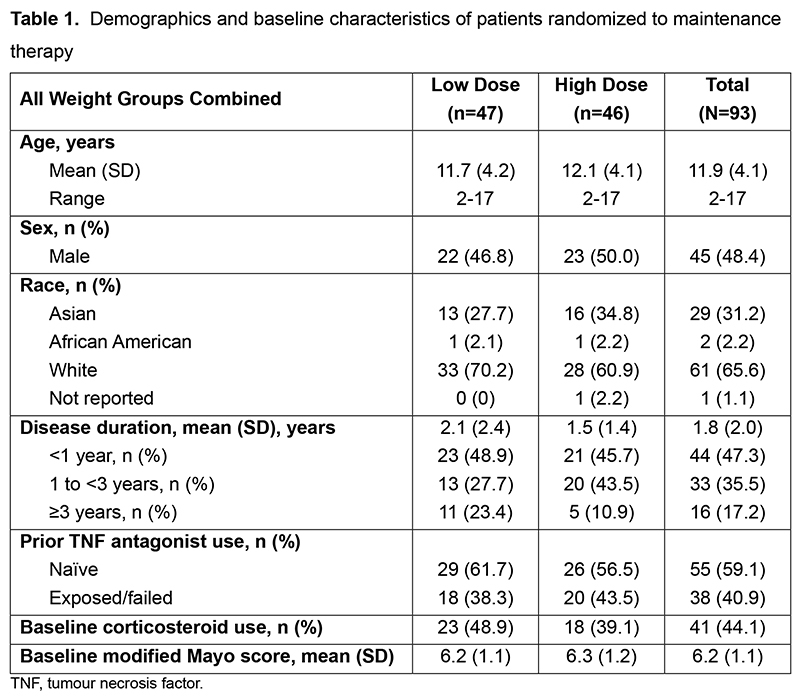
试验共纳入121例患者(10-15kg,n=3;>15-<30kg,n=27;≥30kg,n=91),其中120例接受给药;93例被随机分入LD组(n=47)或HD组(n=46)组开展维持治疗(表)。
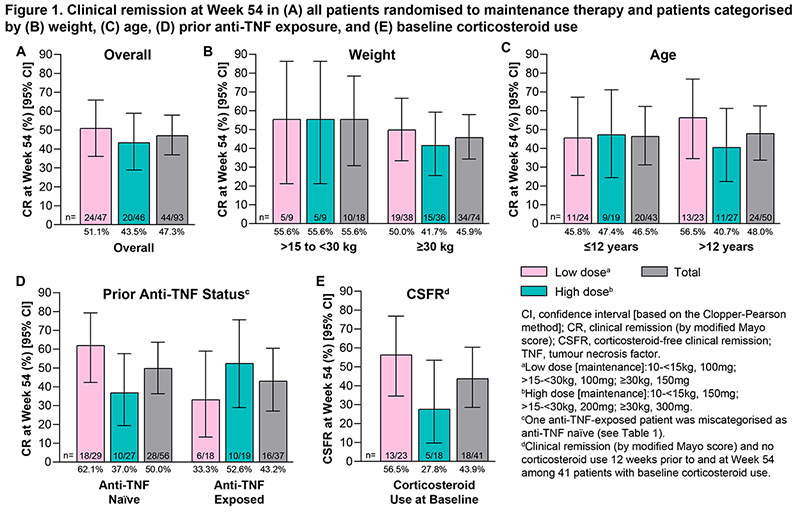
93例患者中44例(47.3%)在第54周达到主要终点(临床缓解),LD组和HD组的比例相近(图1A)。按体重、年龄、抗TNF药物暴露史和基线皮质类固醇使用情况分层的患者在第54周的临床缓解率见图1B-E。
121例患者中42例(34.7%)在第14周达到临床缓解。在第54周,93例患者中27例(29.0%)实现持续临床缓解。在120例暴露于VDZ的患者中,7例(5.8%)出现抗VDZ抗体,其中4例(3.3%)为中和抗体;各体重组间VDZ血清谷浓度相似。
在安全性人群中,120例患者中103例(85.8%)发生治疗期出现的不良事件(TEAE),其中15/120例(12.5%)与VDZ相关。468例TEAE中25例(5.3%)为严重不良事件。5例(4.2%)患者发生与VDZ相关的严重TEAE。7例(5.8%)患者因TEAE导致停用VDZ(最常见:UC恶化,n=3;感染,n=3)。
LD和HD VDZ IV治疗对儿童和青少年UC患者(包括既往抗TNF或皮质类固醇治疗失败者)均有效。在成人UC患者中也观察到了相似的结果。VDZ耐受性良好,未发现新的安全信号。
研究五:UNITI JR研究:乌司奴单抗治疗儿童克罗恩病患者的安全性和有效性结果
(Presentation Number: 404)
在IM-UNITI项目中,乌司奴单抗治疗诱导并维持了成人中重度CD患者的应答和缓解。UNITI Jr研究评估了乌司奴单抗在中重度活动性CD儿童患者中的疗效和安全性。
101例患者(≥2岁–<18岁;pcdai评分>30分;对常规/生物制剂治疗应答不佳/不能耐受或皮质类固醇依赖)接受1次开放标签静脉注射乌司奴单抗(诱导)。在第8周,97例患者按基线体重(<40kg/≥40kg)和应答状态(应答:PCDAI下降≥12.5分且总PCDAI评分≤30分;无应答:PCDAI下降<12.5分)随机分配(1:1),接受盲法皮下乌司奴单抗维持治疗,每8周或每12周(Q8W/Q12W)给药一次,持续44周。乌司奴单抗给药剂量基于体表面积(<40kg的患者)或体重分层(≥40kg的患者)制定。
主要终点是第8周的临床缓解(PCDAI≤10),以及随机化后诱导应答者第52周的临床缓解。分析采用改良意向性治疗(mITT)(仅包括在随机化前第8周应答的患者)。
在UNITI Jr研究中,共纳入101例患者(59.4%男性;中位年龄14.0岁;42.6%未接受生物制剂治疗;中位PCDAI评分40.0);在第8周时,47例患者(46.5%)达到临床缓解。
在第8周达到临床缓解的患者中,32/47例(68.1%)在第52周维持了临床缓解。在第8周时,85例患者(84.2%)达到临床应答,在第52周时,其中46例(54.1%;95% CI:43.6%–64.3%)达到临床缓解,45例(52.9%;95% CI:42.4%–63.2%)达到无皮质类固醇临床缓解。
既往无生物制剂失败史与第52周缓解率更高相关。从第8周到第52周,各体重亚组的缓解率相似。在维持治疗期间,Q8W组和Q12W组的疗效相似。Q8W组与Q12W组的不良事件(AE)/严重不良事件(SAE)发生率相似。16.8%(17/101)的患者发生SAE,其中CD恶化(5/17)最常见;5.9%发生严重感染。
乌司奴单抗耐受性良好,未发现新增安全信号。乌司奴单抗诱导和维持治疗在中重度儿童CD中有效。
 我要投稿
我要投稿















{kind=link}