


1胰腺癌的分子机制
RAS蛋白超家族在细胞信号转导中发挥核心作用,调控细胞增殖、分化及存活等关键生理过程。该家族主要包括KRAS、HRAS(Harvey大鼠肉瘤病毒癌基因同源物)和NRAS(
活性氧作为高活性氧分子,既可损伤生物大分子,亦可介导氧化还原信号通路,通过对靶蛋白的翻译后修饰调控细胞功能。突变型KRAS通过促进线粒体活性氧生成,破坏氧化还原稳态,进而推动肿瘤发生。
丝裂原活化蛋白激酶(MAPK)/细胞外信号调节激酶(ERK)通路是RAS的关键下游效应器。胰腺导管腺癌(PDAC)进展中的高水平ERK激活受其他因子调控。鉴于ERK在KRAS突变胰腺癌中的核心作用,深入解析该通路具有重要研究价值。
KRAS原癌基因
KRAS点突变谱具有组织特异性差异。KRAS原癌基因甘氨酸12→半胱氨酸突变(KRASG12C):占肺腺癌KRAS突变型的40%,但在PDAC中罕见(约1.5%);KRASG12D在PDAC中高发(约40%)。分子机制研究表明,KRASG12C肿瘤主要依赖MAPK信号传导维持恶性表型,故对MAPK抑制剂高度敏感;KRASG12D肿瘤则优先依赖PI3K-Akt-mTOR(哺乳动物雷帕霉素靶蛋白)通路,对该通路抑制剂更易感。联合抑制PI3K-Akt-mTOR通路与KRASG12D可协同诱导癌细胞毒性反应。
除MAPK通路外,KRAS还可有效激活PI3K信号通路。活化的KRAS直接结合PI3K催化亚基p110,诱导其向细胞膜募集并催化磷脂酰肌醇二磷酸转化为磷脂酰肌醇三磷酸。磷脂酰肌醇三磷酸作为第二信使,招募并激活Akt(蛋白激酶B)与PDK1(3-磷酸肌醇依赖性蛋白激酶1)。激活的Akt通过磷酸化下游靶点(如雷帕霉素靶蛋白复合物1、糖原合成酶激酶3β、叉头框蛋白O转录因子和BCL2关联死亡促进因子等),调控细胞存活、代谢、蛋白质合成与生长,在KRAS驱动的肿瘤发生中与MAPK通路同等重要,且两者常存在交互作用(图1)。
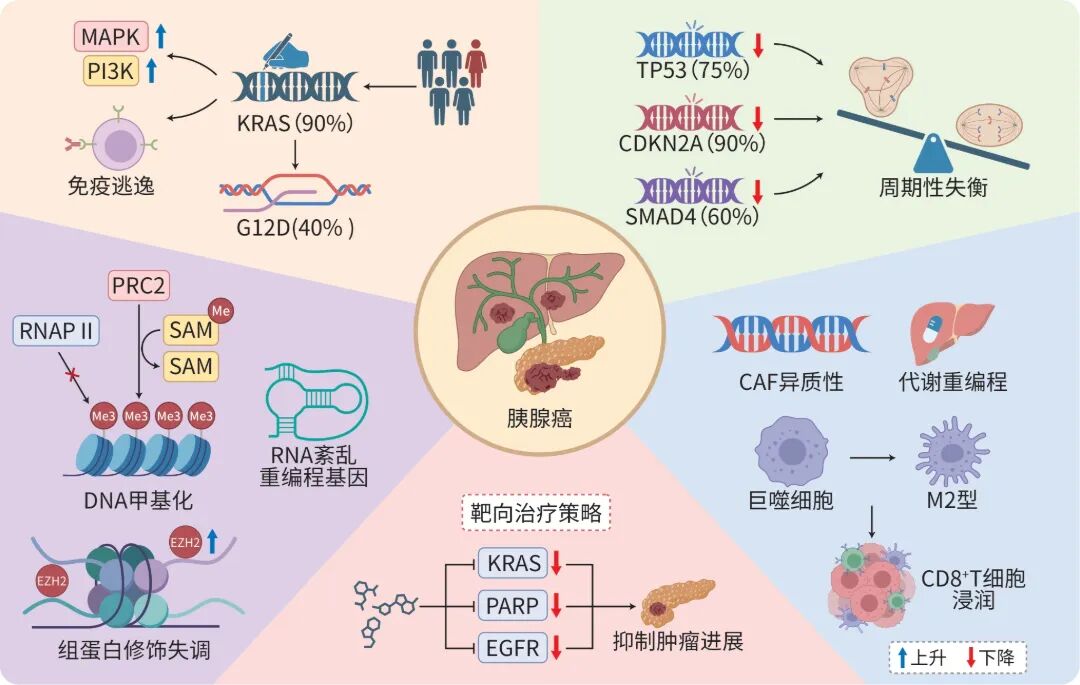
注: MAPK,丝裂原活化蛋白激酶;PI3K,磷脂酰肌醇3激酶;KRAS,Kirsten大鼠肉瘤病毒癌基因同源物;TP53,肿瘤蛋白p53;CDKN2A,细胞周期蛋白依赖性激酶抑制因子2A;SMAD4,SMAD家族成员4;PARP,多
图1 胰腺癌发病机制图
KRAS突变不仅显著调控肿瘤细胞增殖与存活,更通过诱导免疫逃逸、促进血管新生及重塑肿瘤基质,系统性改造肿瘤微环境(TME),加速肿瘤进展。
除KRAS突变外,TP53、CDKN2A和SMAD4等基因的失活同样驱动胰腺癌发生发展。这些改变通过调控细胞周期进程、破坏基因组稳定性、重塑TME,共同促进肿瘤恶性表型(包括侵袭、转移及化疗耐药)的形成(图1)。
TP53是关键的抑癌基因,其编码的p53(肿瘤蛋白p53)通过协调细胞周期阻滞、DNA修复、凋亡及衰老等过程维持基因组稳定性,决定细胞应激命运。然而,约50%的肿瘤存在TP53功能失活,且多数为DNA结合域热点区的错义突变。此类突变赋予p53获得性致癌功能:通过加速糖酵解、促进脂质合成与增强
在PDAC中,50%~75%患者存在TP53突变。最常见突变亚型为p53R273H(小鼠同源物p53R270H),其次为p53R175H(小鼠同源物p53R172H),二者介导差异化的代谢重编程:p53R273H/R270H通过调控线粒体代谢与氧化还原稳态相关基因,驱动代谢紊乱;p53R175H/R172H主要激活
TP53突变是胰腺癌治疗的主要挑战,在晚期胰腺癌中高发,导致不良预后和治疗耐药。突变使
CDKN2A基因(
SMAD4是TGF-β信号通路的核心转导分子,其中非SMAD通路可通过PI3K-Akt信号通路来拮抗SMAD4依赖性的肿瘤抑制,促使TGF-β的功能由抑癌向促癌转换。约60%的胰腺癌存在SMAD4基因失活(突变或纯合缺失),导致其抑癌功能丧失。此外,SMAD4通过调控多能性因子表达维持癌症干细胞(CSC)特性,为靶向治疗提供新方向。
晚期胰腺癌的SMAD4缺失提示高侵袭性与不良预后,其机制包括:(1)直接促癌效应:加速肿瘤增殖;(2)微环境重塑:改变胞外基质成分→促进侵袭转移;(3)免疫逃逸:抑制抗肿瘤免疫应答。三者协同驱动疾病进展恶化。
表观遗传学通过非DNA序列改变调控基因表达。胰腺癌的特征性改变包括:DNA甲基化异常、组蛋白修饰失调和非编码RNA紊乱。这些改变既驱动肿瘤基因表达重编程,又为诊断标志物、预后分层及靶向治疗提供新策略(图1)。
DNA甲基化是表观遗传调控的核心机制,主要发生于CpG岛的胞嘧啶残基,由DNA甲基转移酶催化。该过程通过调控基因表达、维持基因组稳定性以及决定细胞分化命运等方式发挥关键的生物学功能。去甲基化过程则由TET(甲基胞嘧啶双加氧酶)家族介导,通过氧化5-甲基胞嘧啶生成5-羟甲基胞嘧啶实现。
胰腺癌癌前病变——胰腺上皮内瘤变中已存在启动子区异常甲基化,表明表观遗传改变发生于肿瘤早期阶段。异常DNA甲基化作为PDAC的分子标志物,具有分期及亚型特异性,为临床诊断提供重要依据。
组蛋白修饰调控染色质结构与功能,是表观遗传调控的关键机制。在胰腺癌中,组蛋白乙酰化、甲基化及磷酸化等异常修饰驱动恶性进展,具体方式包括:染色质构象重塑影响基因可及性、癌基因异常激活和抑癌基因沉默。组蛋白赖氨酸乙酰化由组蛋白乙酰转移酶催化,通常激活基因转录;其模式异常可显著影响肿瘤生物学行为及治疗反应性。相反,HDAC(组蛋白脱乙酰酶)移除乙酰基,诱导染色质凝集并抑制基因表达。HDAC在PDAC中过表达可沉默抑癌基因,驱动肿瘤进展。
组蛋白甲基化[如组蛋白H3第9位赖氨酸三甲基化(H3K9me3)和组蛋白H3第27位赖氨酸三甲基化(H3K27me3)]是基因沉默的关键表观遗传标记。在胰腺癌中,其异常修饰可导致抑癌基因启动子沉默,具体机制主要涉及以下两种关键酶的作用:果蝇Zeste基因增强子同源物2作为多梳抑制复合物2的催化亚基,能够介导H3K27me3修饰,其过表达与PDAC晚期分期及不良预后显著相关;此外,常染色质组蛋白赖氨酸甲基转移酶2的异常活化可催化H3K9me2修饰,同样提示患者预后较差。这些异常的组蛋白甲基化修饰通过抑制抑癌基因的功能,协同促进胰腺癌细胞的增殖、侵袭转移及耐药性。
非编码RNA,主要包括miRNA(微小RNA)和lncRNA(长链非编码RNA),通过精细调控基因表达深度参与胰腺癌的发生发展。其核心机制如下:miRNA(长度约22 nt)可结合靶mRNA并诱导其降解或翻译抑制,从而精确调控基因表达;在PDAC中,许多miRNA的表达都有明显的异常,如miRNA-21、miRNA-155和miRNA-34a等。lncRNA(如HOX转录反义基因间RNA、转移相关肺腺癌转录本1、HOXA远端转录反义RNA)则在肿瘤生长、上皮-间质转化、代谢重编程、转移及耐药等多个生物学过程中发挥重要调控作用。非编码RNA的多靶点调控特性使其具有治疗潜力,但其组织特异性较低限制了临床转化应用。因此,深入解析lncRNA的作用机制并提升靶向特异性,已成为胰腺癌精准治疗领域的重要研究方向。
miRNA与lncRNA在胰腺癌中并非孤立作用,而是通过复杂的交互形成协同调控网络。其典型模式为:lncRNA可作为竞争性内源RNA(ceRNA)吸附miRNA,从而解除miRNA对其靶mRNA的抑制作用,最终促进癌基因表达并驱动肿瘤的发生与进展。
PDAC的TME高度复杂且异质,特征为多样化细胞组分及可溶性因子间的交互作用。癌症相关成纤维细胞(CAF)存在功能异质性亚群:肌成纤维样CAF、炎性CAF和抗原提呈型CAF。其关键机制涉及诱导M2巨噬细胞极化,从而抑制CD8+T细胞浸润并塑造免疫抑制性TME(图1)。
PDAC的TME富含细胞因子和趋化因子,通过介导免疫-基质细胞交互强化免疫抑制。同时,肿瘤细胞代谢重编程(以葡萄糖代谢异常和缺氧适应为特征)加剧免疫抑制:一方面直接抑制免疫细胞的活化和功能,另一方面创造了有利于肿瘤生长的条件。综上,PDAC-TME通过细胞互作和代谢重塑双重机制,协同促进免疫逃逸和治疗抵抗。

2胰腺癌的靶向治疗策略
靶向治疗凭借其高选择性及低毒性优势,已成为胰腺癌治疗的关键策略。本文对胰腺癌靶向治疗策略的几个主要方向进行概括性总结。
KRAS突变作为PDAC的核心驱动因子(约90%患者携带),其靶向治疗近期取得重要突破。首先,KRASG12C抑制剂(如Sotorasib、Adagrasib和LY3537982)已获批临床应用,显著改善了部分患者的生存预后。此外,针对在PDAC中更常见的KRASG12D突变,MRTX1133通过特异性抑制其GTP酶活性,在肺癌、胰腺癌及结直肠癌模型中展现出强大的抗肿瘤效应。该药物的作用特征包括:高亲和力结合KRASG12D、有效抑制pERK/pS6信号通路及细胞增殖,并在异种移植模型中诱导剂量依赖性的肿瘤消退。尤其在PDAC模型中观察到pERK活性被完全抑制,凸显了其巨大的治疗潜力。
除共价抑制剂外,研究人员正致力于开发非共价抑制剂,旨在持续抑制突变KRAS下游信号通路。最新研发的单克隆抗体12VC1对KRASG12V(KRAS甘氨酸12缬氨酸突变体)和KRASG12C突变体的选择性较野生型KRAS高400倍。该抗体通过有效阻断ERK磷酸化,显著抑制多种KRAS突变细胞系(包括H358人非小细胞肺癌细胞系、PATU8902人胰腺导管腺癌细胞系、HPAF-Ⅱ人胰腺腺癌细胞系和A375人恶性黑色素瘤细胞系)的细胞增殖。尽管靶向KRAS突变的基因治疗在临床前及临床试验阶段已展现出良好前景,但克服关键挑战仍是其成功转化为临床应用的核心所在。
厄洛替尼作为美国食品药品监督管理局批准用于非小细胞肺癌和胰腺癌治疗的药物,通过抑制EGFR激酶活性及其下游信号通路发挥抗肿瘤作用。此外,基于CRISPR的筛选显示,赖氨酸去甲基化酶3A通过KLF5(Krüppel样因子5)和SMAD4调控EGFR表达;厄洛替尼对EGFR的抑制不仅可重塑免疫微环境,还能增强肿瘤对联合免疫治疗的敏感性,提示EGFR抑制剂或可提升“冷肿瘤”PDAC的免疫应答。类似地,拉帕替尼作为另一种酪氨酸激酶抑制剂,可靶向调控的EGFR,通过结合其胞内ATP位点抑制激酶活性,阻断下游信号传导,从而抑制癌细胞异常增殖。然而,当前厄洛替尼的研究存在局限:其作用主要集中于EGFR,对胰腺癌患者(尤其携带KRAS突变者)的临床获益有限。尽管存在不足,厄洛替尼与其他药物(如染料木黄酮)联用可能通过协同效应提升疗效。NCIC CTG Ⅲ期试验(NCT00147497)证实,与吉西他滨单药相比,厄洛替尼联合吉西他滨显著延长晚期胰腺癌患者中位生存期(6.2个月 vs 5.9个月,HR=0.82,P=0.038),并将1年生存率提高至23%。该结果确立了厄洛替尼在晚期胰腺癌一线治疗中的地位。2024年临床前研究表明,厄洛替尼联合线粒体靶向泛醌可显著促进胰腺癌细胞凋亡(凋亡率提升3倍),其协同机制涉及氧化应激通路调控,Ⅰ期临床试验正在筹备。未来研究应更聚焦于临床应用,优化联合治疗策略,以期在胰腺癌治疗领域取得突破性进展。
在DNA修复靶向治疗领域,针对同源重组(HR)缺陷的治疗策略近年来在PDAC治疗中取得重要进展。研究表明,携带BRCA1/2基因突变的PDAC患者对多腺苷二磷酸核糖聚合酶(PARP)抑制剂(如奥拉帕利和尼拉帕利)具有显著治疗反应。该类药物通过抑制DNA单链断裂修复,导致损伤累积,最终诱导癌细胞死亡。该策略尤其适用于存在BRCA突变或其他HR相关缺陷的患者,其抗癌疗效的提升正是基于对肿瘤细胞内在DNA修复缺陷的精准利用。
此外,瑞戈非尼等药物可通过干扰HR修复过程,促进DNA损伤的累积。其作用机制在于抑制关键DNA修复蛋白Rad51的表达,从而削弱PDAC细胞的DNA修复能力,最终增强肿瘤细胞对DNA损伤的敏感性。
除DNA修复靶向治疗外,免疫治疗作为极具吸引力的胰腺癌治疗策略,已展现出潜在价值。靶向程序性细胞死亡受体1(PD-1)及其配体(PD-L1)的免疫检查点抑制剂是当前癌症免疫治疗的主要手段之一。然而,尽管PD-L1抑制剂在多种癌症中疗效显著,其单药治疗PDAC的效果仍不理想。
近年来的临床前研究表明,免疫联合治疗可显著增强PD-L1阻断的疗效。有报告指出,JQ1与PD-L1抗体联用可协同抑制PDAC生长。这些发现表明,整合不同疗法或药物有望克服单一免疫治疗的局限性,从而提升PDAC疗效。优化胰腺癌免疫疗法需深入解析PD-L1表达调控机制并识别影响免疫治疗应答的生物标志物,从而为患者制订个体化治疗方案。
免疫治疗的效果常因肿瘤产生内在或获得性耐药而受到限制,这反映出当前对免疫压力下肿瘤演化机制的理解尚不充分。CSC可通过多种途径逃避免疫监视,例如低表达主要组织相容性复合体分子、分泌免疫抑制性细胞因子,以及募集调节性T细胞和髓源性抑制细胞等。在免疫治疗的选择压力下,这类具有免疫特权特征的CSC能够迅速扩增并分化,形成具有时空连续性的耐药群体。该过程不仅依赖于CSC自身的固有特性,也与其和TME中多种细胞成分的动态交互密切相关,通过黏附分子、配体-受体信号等机制调控免疫细胞与基质细胞的功能,共同促进免疫逃逸和肿瘤存活。
有研究显示,miR4435-2宿主基因通过miR-1252-5p/STAT1调控轴促进胰腺癌干性特征、肿瘤进展及化疗耐药,发挥促癌作用。上述发现为靶向miRNA调控CSC提供了新的治疗策略。

3结论与展望
胰腺癌恶性程度高且早期诊断困难,其治疗始终是临床医学面临的重大挑战。近年来,随着分子机制研究的深入及靶向治疗策略的进展,为胰腺癌治疗带来了新机遇。KRAS突变、EGFR信号通路、DNA修复机制等已成为关键治疗靶点。厄洛替尼、拉帕替尼等靶向药物的研发与应用,为临床提供了有效选择。联合治疗,尤其是靶向治疗与化疗、免疫治疗的结合,已成为临床研究的前沿,为胰腺癌治疗开辟了新思路。
尽管靶向治疗已取得突破性进展,但胰腺癌因其高度异质性、耐药性及免疫逃逸等独特难点,治疗仍面临严峻挑战。未来研究需深入解析胰腺癌的分子机制,特别是KRAS突变及其下游信号通路,以及独特的肿瘤免疫微环境,以期发现更多潜在靶点,为靶向治疗拓展新方向。精准医疗理念日益凸显,基于患者分子特征制订个体化治疗方案将得到更广泛应用,从而提高疗效并降低副作用。优化联合治疗策略是重点,例如高效组合靶向药物、免疫治疗与化疗药物,有望克服耐药性、减少不良反应并增强抗肿瘤效果。此外,针对胰腺癌特有分子特征的新靶点探索及新药研发,也将是重要的研究方向。

https://www.lcgdbzz.org/cn/article/doi/10.12449/JCH260234

刘铁鑫, 郭松雨, 王震侠. 胰腺癌发生发展的分子机制与靶向治疗策略[J]. 临床肝胆病杂志, 2026, 42(2): 490-496
来源:临床肝胆病杂志
本平台旨在为医疗卫生专业人士传递更多医学信息。本平台发布的内容,不能以任何方式取代专业的医疗指导,也不应被视为诊疗建议。如该等信息被用于了解医学信息以外的目的,本平台不承担相关责任。本平台对发布的内容,并不代表同意其描述和观点。若涉及版权问题,烦请权利人与我们联系,我们将尽快处理。
 我要投稿
我要投稿













{kind=link}