编者按
新生儿期出现的紧张性水疱大疱,组织病理显示表皮下大疱伴大量嗜酸性粒细胞浸润——这一组合常指向自身免疫性大疱病,如大疱性类


主诉
新生儿因胸、四肢出现
现病史
患儿入院前1天无明显诱因于胸部、双上肢、双下肢出现约15个散在分布的水疱、大疱,大小2~10mm,呈黄色,兼具张力性与松弛性表现;伴轻微屈侧红斑(臀沟、腋窝右侧重于左侧),无明显皮肤触痛、口周糜烂、鳞屑表现。患儿无
既往史
既往有新生儿
家族史
家族中无类似皮肤病及遗传性疾病史,无特殊疾病遗传背景。
体格检查
胸、四肢见散在2~10mm黄色张力性/松弛性水疱、大疱,屈侧(臀沟、右腋窝)轻度红斑(图1A,原文左侧上肢临床影像);无发热,全身一般情况良好,未查见其他系统异常体征。
3个月随访查体:仍可见水疱、糜烂,伴色素减退斑、色素沉着斑;甲
1年随访查体:面部、全身见棕褐色斑片,呈斑驳状色素沉着表现。
辅助检查
1.组织
2.免疫组化检查:Ⅳ型胶原免疫组化(×200,图2)示疱底可见阳性标记。
3.免疫相关检查:直接免疫荧光、
4.免疫表型检测:角蛋白14(KRT14)、角蛋白5(KRT5)表达弱阳性,网蛋白染色微量阴性。
5.基因检测:高通量测序示KRT5基因杂合致病性变异p.P25L,该变异在基因组聚合数据库中未收录,归为常
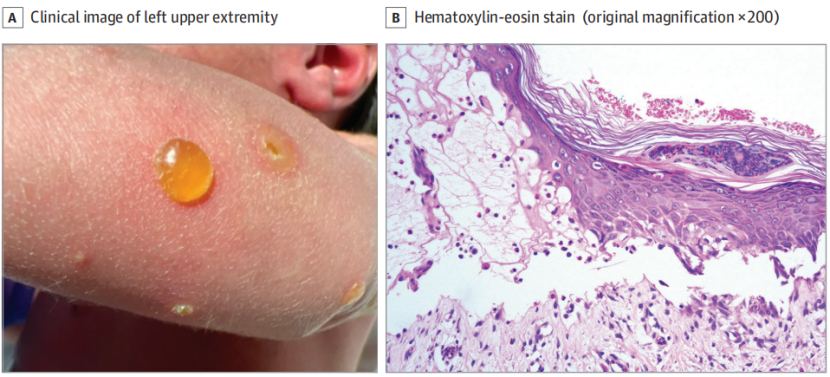
图1 左上肢可见散在分布的2~10mm张力性与松弛性黄色水疱、大疱;B:表皮下大疱形成,疱腔及乳头真皮内可见大量嗜酸性粒细胞。
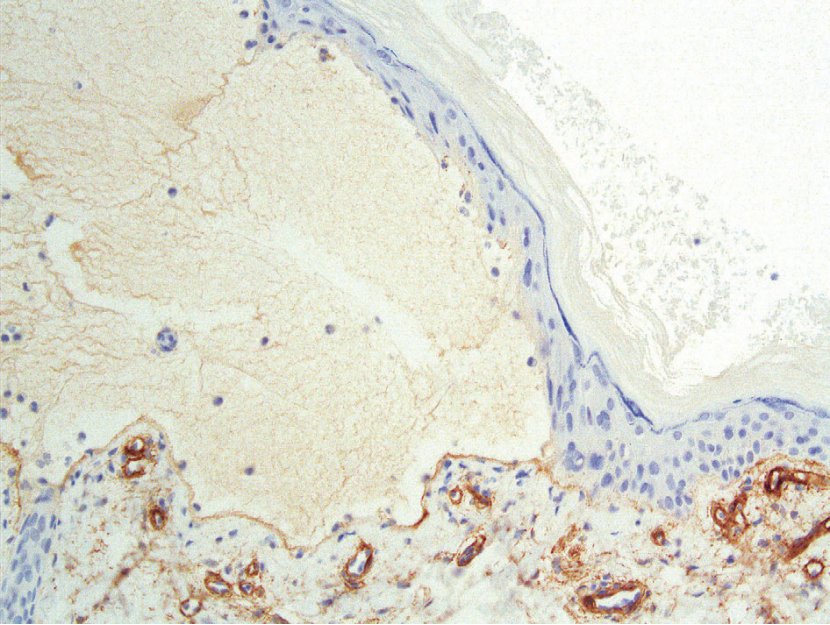
图2 Ⅳ型胶原免疫组化染色(原始放大倍数×200),可见疱底呈阳性标记。
诊断与鉴别诊断
最终诊断
色素斑驳型单纯性大疱性表皮松解症(EBS),常染色体显性遗传(KRT5基因p.P25L杂合突变)。
鉴别诊断
1.大疱性类天疱疮(BP):虽可表现为表皮下大疱、嗜酸性粒细胞浸润,且张力性大疱为典型表现,但本病在婴儿中罕见;且本例患儿及母亲均无BP180、230抗体,直接免疫荧光无真皮-表皮交界处线性IgG和C3沉积,疱底基底层细胞剪切样改变也与BP不符,可排除。
2.大疱性脓疱疮:患儿水疱呈黄色,需与本病鉴别;但大疱性脓疱疮组织病理为角层下大疱,疱内可见中性粒细胞及革兰染色阳性细菌,与本例表皮下大疱、嗜酸性粒细胞浸润的病理特征不符,且无感染相关临床及病理依据,可排除。
3.大疱性超敏反应:可出现表皮下大疱伴嗜酸性粒细胞浸润,但本病皮疹多在诱因去除后消退,无持续性色素异常及甲营养不良表现,与本例病程中持续的色素斑驳、甲改变不符,可排除。


色素斑驳型单纯性大疱性表皮松解症是EBS的罕见亚型,为KRT5或KRT14基因变异导致的常染色体显性遗传性皮肤病(偶为隐性);上述角蛋白构成的中间丝网络是基底层角质形成细胞锚定于基底膜的关键结构,基因变异会破坏细胞骨架稳定性,使基底层细胞易受轻微外伤发生溶解,临床以皮肤脆性增加、网状色素沉着、掌跖角化、甲营养不良为核心特征。
本例诊断的核心难点在于遗传性皮肤病表现出自身免疫性大疱病的组织病理特征:经典EBS多为表皮内(基底层内)裂隙,炎症细胞稀少,伴松弛性大疱;而色素斑驳型EBS在炎症性
本例的关键诊断线索为疱底基底层细胞的轻微剪切样改变,提示裂隙平面更表浅,与EBS的病理本质相符;而免疫荧光(排除自身免疫性病因)、免疫组化(明确裂隙平面)及基因检测(确诊KRT5基因突变)则为最终诊断提供了决定性依据。临床中当组织病理结果与临床怀疑不符时,免疫荧光定位、角蛋白表达检测及分子基因测序是鉴别
误诊为大疱性类天疱疮可能导致婴儿接受不必要的免疫抑制治疗,带来显著临床风险;而EBS的精准诊断可为临床制定避免外伤、创面护理的对症治疗方案及家族遗传咨询提供依据。EBS的水疱表现多随病程逐渐改善,但色素异常和甲改变可能持续存在;目前本病尚无根治方案,激酶抑制剂米哚妥林在患者来源的EBS角质形成细胞模型中可改善细胞骨架稳定性,相关角质聚集调节剂、炎症通路抑制剂的研究仍在进行中。


本病例提示,新生儿以张力性水疱大疱伴嗜酸性粒细胞丰富的表皮下大疱为表现时,需警惕色素斑驳型EBS的可能,疱底基底层细胞剪切样改变是重要的病理线索;临床需整合临床表现、组织病理、免疫病理及分子遗传学检查结果,才能有效鉴别遗传性大疱性表皮松解症与获得性自身免疫性大疱病,避免误诊误治,为患者制定合理的治疗及随访方案。
参考文献:
医脉通是专业的在线医生平台,“感知世界医学

 我要投稿
我要投稿















{kind=link}