62岁女性出现进行性痉挛、无力和步态不稳,该如何诊断?| 临床推理
2025-07-31
来源:医脉通
成人发病的步态不稳在临床诊断中存在一定挑战。近期,Neurology杂志上报告了一例62岁女性步态不稳的鉴别诊断过程。该患者表现为慢性进行性下肢无力、感觉障碍和步态不稳,随后出现认知困难、尿急和夜间视力受损。患者既往具有手术矫正的锤状趾病史。本病例强调了通过构建全面的鉴别诊断来诊断步态不稳的重要性,特别关注了神经定位并探索了各种可能的病因,为步态不稳患者的临床诊断提供了参考。
一名62岁女性,具有高血压史,表现为进行性步态不稳和下肢无力,病程持续进展至少10年。
患者40多快50岁时出现锤状趾,此前未出现任何下肢感觉运动缺陷,且在童年时期达到所有发育里程碑。50岁时,被诊断为腓骨肌萎缩症(CMT),一年后接受脚趾矫正手术。55岁左右,出现走路双腿交叉,导致经常摔倒。50多快60岁时,出现双足麻木和无力,需间歇性使用手杖。患者否认自主神经功能障碍史,包括体位性低血压。无神经系统疾病家族史。
初步神经系统检查显示脑神经完整,患者视野、瞳孔反射、眼球和面部运动均正常,面部感觉正常且对称,无眼球震颤、构音障碍或明显的下颌反射。患者上肢近端和远端表现出轻度对称性无力,主要影响手的伸肌和固有肌;下肢近端表现出轻度无力,远端表现出中度无力,主要影响屈肌。患者手部、小腿和足部远端固有肌萎缩,且具有高弓足和手术矫正后的锤状趾;患者髋关节肌张力增加,踝关节肌张力降低,小腿中部失去对温度和针刺的感觉,同时双侧大脚趾的震动和本体感觉减弱;全身深部肌腱反射增强,但踝关节处缺失;双侧足底反射为伸肌反射;步态狭窄,双腿交叉。Romberg征阳性。无明显躯干共济失调或肢体共济失调。
随后的评估中,患者步态和平衡能力逐渐恶化,至50多岁时需使用助行器,60岁出头即需轮椅。患者50多快60岁时,出现认知困难、尿急和夜间视力受损,但神经心理学测试显示未达到轻度认知障碍或痴呆的标准。眼科检查发现视网膜营养不良。
问题思考:
1.该患者临床表现的定位是什么?
2.可能的鉴别诊断有哪些?疾病进展和症状演变如何影响鉴别诊断?
该患者表现为隐性进展的、对称的、远端大于近端的上运动神经元无力,并伴有远端肌肉萎缩和以长度依赖性分布的感觉障碍。随后,患者又出现认知障碍和尿急症状。患者最初表现为一种脊髓神经病变,影响四肢的对称性双侧上运动神经元无力提示脊髓水平的皮质脊髓束功能障碍,而远端肌肉萎缩和以长度依赖性分布的感觉障碍提示伴多发性神经病。
成人脊髓神经病的鉴别诊断包括代谢综合征(如维生素B12/E/铜缺乏症、锌/一氧化二氮中毒、线粒体综合征)、感染综合征(如HIV、HTLV-1/2热带痉挛性截瘫)、遗传综合征(如遗传性痉挛性截瘫、肾上腺脊髓神经病、成人发病的白质营养不良、脊髓小脑共济失调、Friedreich共济失调)。副肿瘤综合征包含于脊髓神经病的鉴别诊断中,但鉴于该患者起病隐匿,考虑副肿瘤综合征的可能性较小。如果感觉功能障碍不明显,考虑运动神经元疾病;如果患者仅表现为慢性进行性脊髓病症状而无神经病变,则考虑脊髓血管异常,如硬脊膜动静脉瘘。
对于该患者而言,慢性且缓慢进展的脊髓神经病变符合大多数遗传性脊髓神经病的时间模式。虽然通常表现为亚急性,但必须排除包括营养、感染和炎症(如中枢神经系统脱髓鞘疾病)在内的其它病因,因为这些病因具有疾病修正干预的潜力。如果多发性神经病和脊髓病之间没有明显的时间相关性,排除脊髓的结构性病变也非常重要,因为这些病变可能表现为慢性或急性伴慢性脊髓病,可模拟脊髓神经病。该患者疾病晚期出现的认知障碍,可能与多种脊髓神经病综合征相关,包括维生素B12缺乏、一氧化二氮中毒、线粒体综合征、HIV及许多遗传综合征。
问题思考:
1. 哪些检查可以帮助缩小鉴别诊断范围?
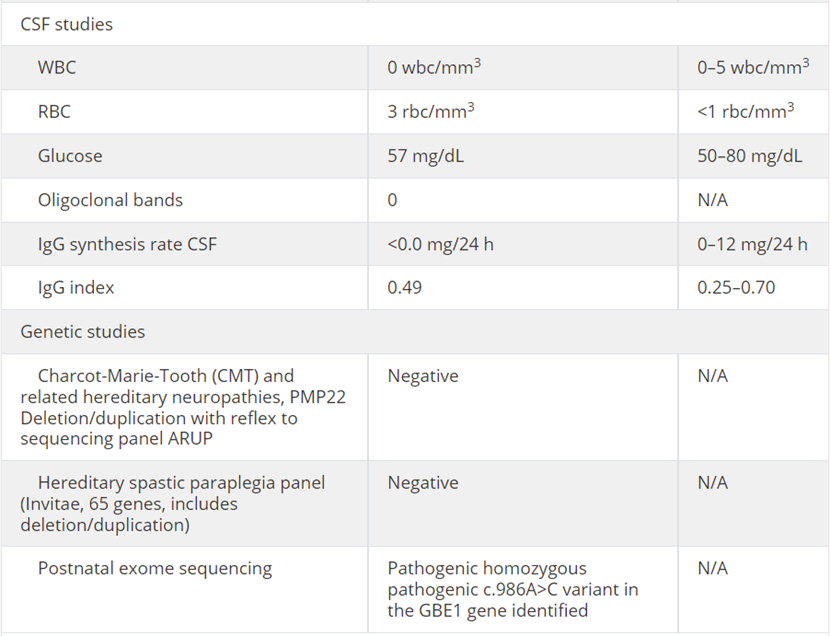
该患者接受了脊髓病及神经病中可获得性和可逆性病因的初步评估。实验室检查筛查了各种可获性代谢病因,包括全血细胞计数(CBC)、完整代谢面板、维生素B12、铜和铜蓝蛋白;炎症病因筛查包括血清红细胞沉降率(ESR)、C反应蛋白(CRP)、抗核抗体(ANA)、抗水通道蛋白4抗体和组织转谷氨酰胺酶抗体;感染病因筛查包括梅毒、HIV和HTLV-1/2血清检测、脊髓液分析,包括细胞计数、葡萄糖、蛋白质、寡克隆带、IgG指数和IgG合成率(见表1)。
表1 实验室检查结果
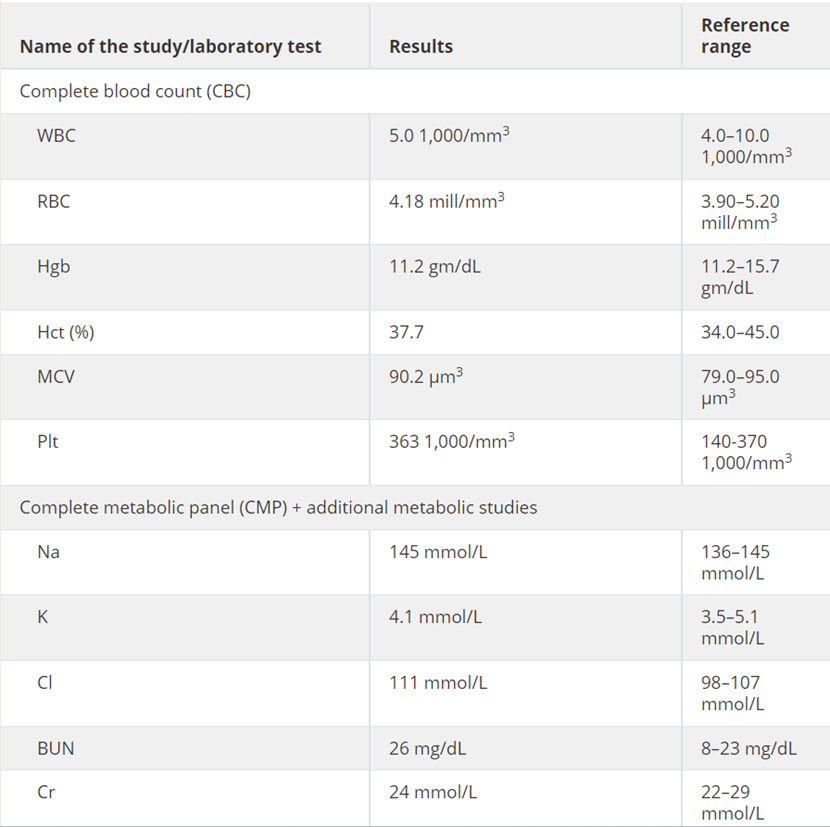
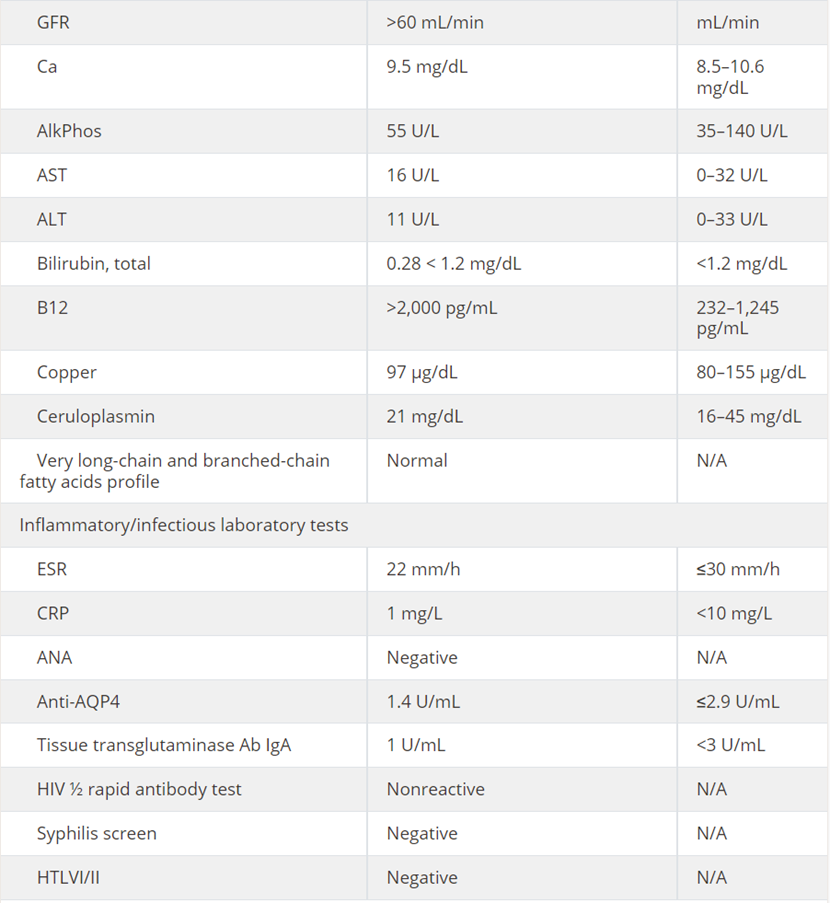
为排除脊髓病和皮质脊髓束功能障碍的结构性和血管性病因,进行了脑部、颈椎和胸椎MRI检查(见图1),结果显示延髓、颈髓和胸髓明显萎缩:(1)延髓前锥体中观察到T2高信号病变,向上延伸至脊髓小脑束、以及楔束和股薄肌内的背侧延髓;(2)背侧脑桥穿过皮质脊髓束延伸至双侧内囊后肢;(3)小脑脚延伸至齿状核。肌电图(EMG)/神经传导速度(NCS)检测显示存在具有轴突特征的长度依赖性感觉运动多发性神经病。
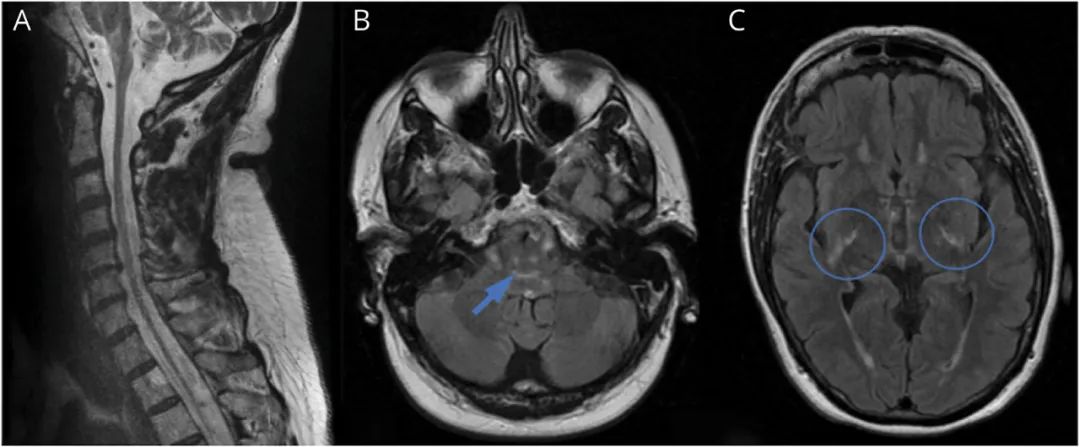
图1 患者脑部和颈椎MRI (A)颈椎矢状面T2加权MRI显示延髓和颈部脊髓严重萎缩。(B和C)轴位T2 FLAIR脑部MRI显示脑桥背柱高强度病变(箭头所示)延伸至内囊后肢(圆圈所示)
随后进行遗传病因评估。值得注意的是,无家族史并不排除遗传病因评估,尤其是在需要两个隐性等位基因的常染色体隐性遗传疾病中。患者首先进行了血清长链脂肪酸水平和遗传性痉挛性截瘫的基因面板检测,结果呈阴性。后接受全外显子基因测序发现GBE1基因中存在纯合子c.986 A>C致病变异。最终该患者被诊断为成人多葡聚糖体病(APBD)。
APBD是一种罕见的常染色体隐性遗传脑白质营养不良病,通常于生命的第五或第六个十年开始发病,多见于拥有犹太血统的人群。截至目前,全球约有200例APBD患者。鉴于该疾病的罕见性,APBD的诊断常常被延误。APBD主要由糖原分支酶(GBE)缺乏导致多葡聚糖体积累,进而干扰中枢和外周神经系统的糖原代谢所引起。这些干扰在患者MRI上清晰可见,尤其在T2加权和FLAIR序列中,表现为高信号病灶与神经系统受累区域(包括室旁、皮层下和深部脑白质改变)中多葡聚糖体的广泛积累相关,涉及上脑桥、延髓、小脑脚和脊髓。患者之间最一致的异常表现为延髓和脊髓萎缩。
APBD的特征是进行性锥体截瘫、下肢远端感觉缺失和神经源性膀胱(通常为典型的早期特征)。根据2012年对50名APBD合并GBE1缺乏症患者的全球临床和实验室研究数据显示,神经源性膀胱的中位发病年龄为51岁,依赖轮椅的中位年龄为63岁,死亡的中位年龄为70岁。定期监测对于观察疾病进展非常重要。约30%的APBD患者为具有异常RNA剪接的异合子病征基因型,因此,未受影响的家庭成员应考虑进行遗传咨询和检测。
APBD的管理主要是对症治疗,需神经科医师、泌尿科医师、物理治疗师等多学科团队合作。最近,愈创木酚作为一种天然调味剂,可通过降低小鼠模型中的多葡聚糖体积累而改善小鼠寿命和握力,被视为APBD的候选治疗药物。
参考文献
1.Voloshyna-Farber EY, Pappo C, Kansal L, Ferrey DA. Clinical Reasoning: A 62-Year-Old Woman With Progressive Spasticity, Weakness, and Gait Instability. Neurology. 2025 Feb 11;104(3):e210290.
(本网站所有内容,凡注明来源为“医脉通”,版权均归医脉通所有,未经授权,任何媒体、网站或个人不得转载,否则将追究法律责任,授权转载时须注明“来源:医脉通”。本网注明来源为其他媒体的内容为转载,转载仅作观点分享,版权归原作者所有,如有侵犯版权,请及时联系我们。)








