病例介绍

一名37岁女性患者因
次年,32岁的患者在
37岁时,患者出现

抗磷脂综合征概述
抗磷脂抗体综合征(APS)是一种目前无法治愈的血栓炎症性疾病,其特征包括静脉和动脉宏观血管血栓事件风险增加、微血管损伤、胎盘功能不全、心脏瓣膜疾病、
此外,抗凝剂无法保护微血管系统,导致部分患者随时间推移出现器官(脑、心脏、肾脏等)功能恶化。目前仍存在对安全有效疗法的需求,以对抗APS的炎症性、抗凝剂抵抗性表现。除了临床表现外,APS的正式分类标准还要求
另外,狼疮抗凝物试验—通过一系列凝血检测来筛选相关自身抗体,其原理是这些抗体能在体外异常延长磷脂依赖性
图1重点展示了临床可用的抗磷脂抗体(aPL)检测方法、其所检测的循环自身抗体与相关临床场景之间的关联。本文将探讨抗磷脂抗体介导的抗磷脂综合征(APS)机制,特别聚焦于近期对抗β2GPI抗体认知的新发现。鉴于APS自身抗体下游存在多种可能协同作用的机制:清除致病性自身抗体是治疗APS患者并最终实现治愈的唯一有效策略。
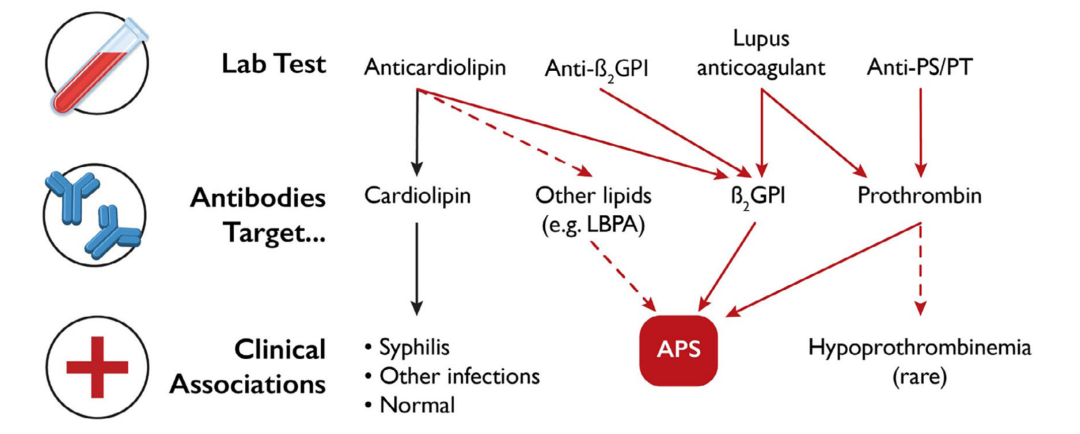
图 1. 抗磷脂抗体检测及与临床关联。该示意图重点展示了常用实验室检测方法、导致其阳性反应的自身抗原以及临床关联之间的关系。虚线表示尚未完全明确的关联。APS,抗磷脂抗体综合征; β2GPI,β -2糖蛋白I; LBPA,溶血双磷脂酸;PS/PT,磷脂酰丝氨酸/凝血酶原
APS病理生理学概述
APS相关自身抗体不仅是疾病状态的生物标志物,更是潜在病理生理学机制的驱动因素。这些抗磷脂抗体(aPL)与磷脂结合,更重要的是与细胞表面的磷脂结合蛋白(如 β2GPI和凝血酶原)结合,从而激活中性粒细胞、血小板、内皮细胞、单核细胞和补体系统——在局部或全身应激时使循环血液趋向血栓形成。
抗体持续循环而血栓事件偶发的现象,催生了APS相关血栓形成的“二次触发假说”。该假说认为,APS患者的循环系统在遭遇环境诱因时,其血栓形成阈值会显著降低。虽然本文不会详述动物模型,但研究者们长期将APS患者分离的全免疫球蛋白G(IgG)组分或亲和纯化IgG组分移植至小鼠(偶尔也包括其他实验动物)体内,以诱发促血栓状态。 与人类情况类似,抗体介导的血栓在动物体内不会自发形成,但当血管受到机械性或炎症性损伤时,血栓形成会显著加剧。
β2GPI在健康和疾病中的作用
如前所述,靶向血浆蛋 白β2GPI(尤其是其结构域I)的抗体具有特别强的致病性。最初被命名为载脂蛋白H(AP-OH)的β2GPI含有五个补体控制蛋白样结构域。与其他结构域不同,结构域V具有独特的阳离子区域和独立的疏水环,共同决定了β2GPI与带负电荷磷脂结合的倾向性。在循环系统中浓度约为200μg/mL的 β2GPI具有明显的稳态特性,包括清除凋亡碎片、抑制过度感染相关炎症信号传导,甚至限制血管生成和癌细胞迁移。
与APS相关的是,β2GPI还可能具有天然的抗血栓特性,尽管作用温和。例如,通过竞争性阻断凝血因子与细胞表面的结合,β2GPI可能在血栓形成背景下抑制过度凝血和血小板活化。尽管 β2GPI在不同物种间高度保守,但似乎并非人类或小鼠健康发育所必需。
历史上,β2GPI被认为存在两种构象:在血液中呈环状,其自身抗原性结构域I与结构域V紧密关联;当与表面结合时则呈开放状态,形似延长的鱼钩。 在此构象中,结构域I仅在蛋白质处于开放构象时可被接触,该构象由表面结合的磷脂触发。大多数细胞表面对 β2GPI的积累相对抵抗,但在应激状态下这一特性会发生改变。有趣的是,胎盘内皮细胞和滋养层细胞似乎是个例外,因为这些细胞表面在基线状态下存在 β2GPI。
尽管该模型解释了循环中未检测到 β2GPI/抗 β2GPI复合物的原因,但近期研究提出了另一种解释。这些新报告表明,β2GPI的伸长形式是循环中的主要形式,且在天然条件下从人血浆中分离的β2GPI具有抗环化特性 。此外,虽然抗 β2GPI抗体可在溶液中结合 β2GPI,但其结合亲和力相对较弱。
综合这些数据,研究者提出一个模型:β2GPI的表面积累通过改变局部 β2GPI的丰度和取向来支持抗 β2GPI的识别,而非通过强力打开蛋白质结构实现。简而言之,抗原-抗体复合物仅当抗体的两个Fab区域各自具有可结合 β2GPI结构域I时才能保持稳定。
近期其他研究对 β2GPI结合的普遍认知提出了挑战。尽管β2GPI最广为人知的功能是结合磷脂,但我们现在知道它还能与游离DNA及中性粒细胞胞外诱捕网(NETs)相互作用,这很可能通过其结构域V中促进磷脂黏附的富含
抗β2GPI形成
尽管目前尚未就β2GPI结构域I耐受性丧失的成因达成共识,但可能存在多种潜在的重叠机制。例如,AP-OH已有研究描述了导致S88N、V247L、C306G和W316S等变异的多态性 。其中,V247L变异与抗磷脂抗体(aPL)发生及相关临床表现风险增加相关。相反,C306G和W316变异被报道可改变血清中β2GPI水平及表面结合能力,从而降低aPL产生的可能性。
β2GPI的翻译后修饰和/或新型蛋白质复合物的形成可能是抗β2GPI形成的因素。在APS患者中,β2GPI的氧化程度增加,尤其是那些抗β2GPI IgG靶向该蛋白结构域I的患者。二硫键C288-C326的氧化增强了β2GPI的表面结合能力,从而支持抗磷脂抗体(aPL)的识别。 氨基甲酰化β2GPI也被认为是aPL的作用靶点。除了翻译后修饰之外,一组研究人员描述了完整的、未经加工的β2GPI与主要组织相容性复合体II类的异常结合,随后这两种蛋白质共同呈现在细胞表面。在APS患者中观察到了针对这种新型复合物的抗体,这些抗体可能促进疾病相关的血栓形成。促炎信号通路抗β2GPI抗体的下游三种抗β2GPI型:IgG、IgM和IgA。
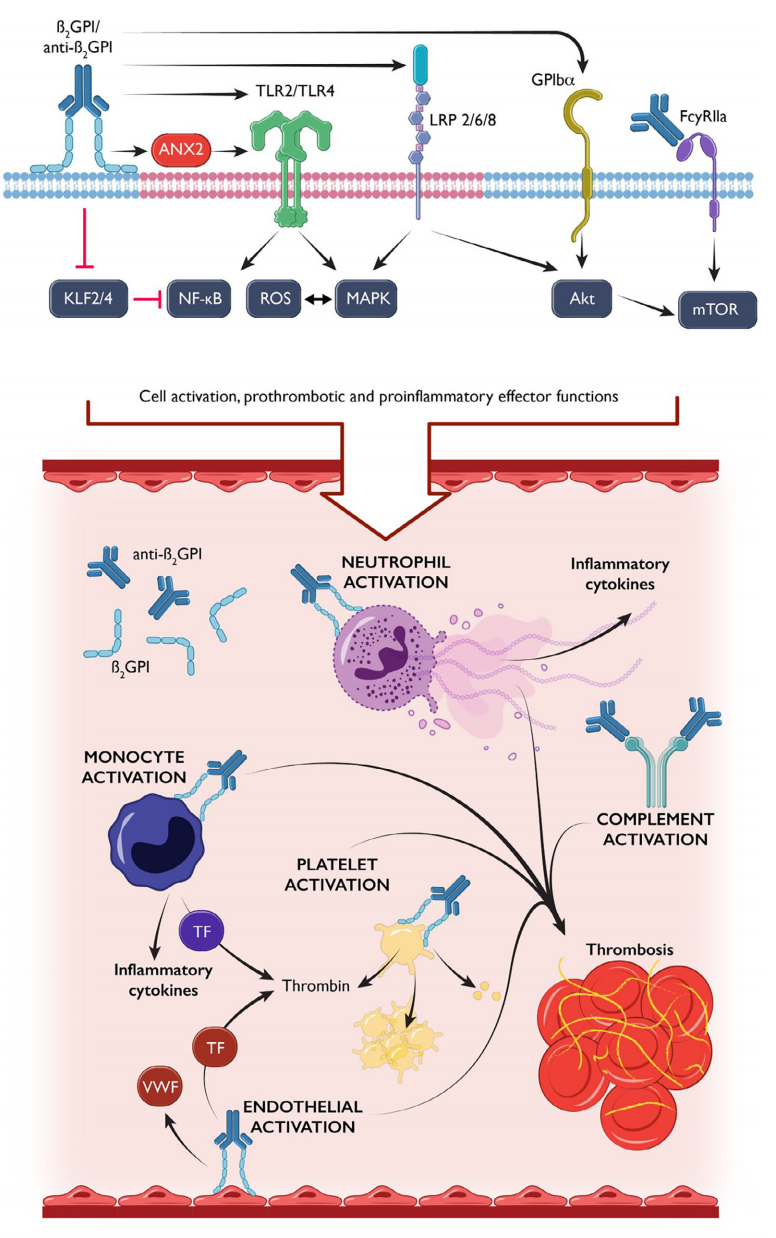
其中,抗β2GPI IgG被认为与APS的诊断和发病机制最为相关。尽管现行分类标准中也考虑了抗 β2GPI IgM,但IgM同种型aPL的功能相关性仍知之甚少,甚至有研究提出此类抗体在系统性红斑狼疮(SLE)背景下可能具有保护作用 。我们对抗β2GPI IgA的认知,尤其是在功能层面,同样存在不足。因此,以下机制几乎完全针对IgG同种型。β2GPI二聚体通过抗β2GPI IgG的结合而稳定,似乎与多种表面共受体相互作用以发挥致病效应(图2)。其中一种因素是膜联蛋白A2,一种通常支持
抗β2GPI激活促血栓形成的机制
中性粒细胞通过释放中性粒细胞胞外诱捕网(NETs)参与APS的发病机制。NETs是由染色质骨架构成的网状细胞外结构,表面装饰着胞质和颗粒来源的蛋白质,这些物质通过中性粒细胞胞外诱捕网形成过程(NETosis)释放。APS相关的NETosis由β2GPI/抗 β2GPI 表面复合物通过TLR4依赖性活性氧生成激活。NETosis过程中释放的多种细胞内因子具有促炎和促血栓形成作用。例如,DNA和组织因子可激活凝血级联反应,而NET来源的蛋白酶则能灭活抗凝酶。NETs中还存在钙卫蛋白等警报素,可能降低血小板活性。NETs促进下游干扰素和其他炎症细胞因子的产生,这可能支持针对细胞外空间中核和其他抗原的自身抗体库的扩张。因此,NETs可能不仅在β2GPI相关血栓形成中发挥作用,而且在推动APS向前发展的自我持续炎症中发挥作用。
血小板在APS病理生理学中也发挥作用,β2GPI/抗β2GPI复合物可能通过血小板衍生的CXCL4(血小板因子4)在血小板表面稳定。 这些表面复合物通过多种受体(包括TLR2、ApoER2、GPIbα 和 FcγRII)激活血小板。受体激活后的下游信号传导(通过雷帕霉素靶蛋白/Akt机制)显得尤为重要。 此外,β2GPI/抗β2GPI也可能通过与中性粒细胞、内皮细胞、单核细胞及补体系统的相互作用间接激活血小板。一旦激活,血小板表现出增强的聚集性、凝血酶生成及微粒释放。
由于血管性APS最常见的临床表现是动脉或静脉特定部位的血栓事件,局部内皮扰动无疑是循环细胞类型参与血栓形成的关键调控因素。β2GPI/抗β2GPI表面复合物在内皮受压时积累,通过TLR和LRP介导的信号通路激活内皮细胞。与此同时,抗炎性Kruppel样因子2和4受到抑制,进一步支持内皮细胞的激活。活化的内皮细胞通过在其表面呈现黏附分子和组织因子,并释放
APS发病机制中的其他因素
除了前述的细胞类型外,单核细胞在血栓事件中也是凝血激活组织因子的关键呈递者,这一过程在APS中进一步加剧。值得注意的是,不同风险特征的APS患者可通过单核细胞基因表达进行区分,且多种类型的抗磷脂抗体(包括非辅因子依赖性抗体)已被证实可激活促血栓单核细胞信号传导。滋养层细胞是另一种易受 β2GPI/抗β2GPI IgG介导激活影响的细胞类型,这对APS常见的胎盘功能不全具有临床意义。
另有证据表明,补体系统在APS中通过Fc依赖性和非依赖性途径异常激活,进一步加剧了细胞活化与损伤,其程度超出单纯表面β2GPI/抗β2GPI介导信号传导所能达到的水平。 最后,抗磷脂抗体可能通过尚未完全阐明的机制以细胞非依赖性方式影响凝血通路,包括触发活化
抗心磷脂免疫检测方法最初是为检测β2GPI依赖性抗心磷脂抗体而优化的。相比之下,辅因子非依赖性抗心磷脂抗体在抗磷脂综合征(APS)发病机制中的作用尚不明确,这也解释了为何
近年来一个新发现是针对溶血双磷酸脂酸与单核细胞、内皮细胞等细胞表面的蛋白C受体复合物的辅因子非依赖性抗体。传统APS检测方案难以识别此类抗体,需要更多独立研究团队开展工作以评估其在临床中的潜在作用。除了最新分类标准中涵盖的aPL之外,有证据表明,通过狼疮抗凝物检测或抗PS/PT免疫分析法检测到的抗凝血酶原IgG抗体(图3),在凝血酶原、钙和 FcγRIIA 依赖性信号存在的情况下激活血小板。
APS治疗的未来方向
维生素K拮抗剂(如华法林)仍是降低APS患者复发性血栓风险(尽管效果不完全)的金标准。在高风险或难治性临床情况下,有时会采用辅助治疗,包括抗血小板药物(如低剂量
尽管部分患者在使用维生素K拮抗剂治疗期间病情相对稳定,但与APS患者的交流很快让我们意识到,这类药物会带来显著的生活质量负担,包括饮食调整、耗时的监测,以及最重要的持续性出血风险。此外,使用抗凝剂治疗自身免疫性疾病这一鲜明矛盾立即显现。目前仍对能够靶向抗磷脂抗体(aPL)介导的凝血上游炎症事件的方法保持持续关注该系统的作用机制包括阻断或拆除NETs、抑制内皮细胞的隐匿性活化,以及破坏炎症放大补体信号传导。然而需要警惕的是,由于不同患者体内可能同时存在多种作用机制,若仅针对单一通路进行干预,将难以实现APS的完全抑制。鉴于体外实验和动物模型系统提供的有力证据表明抗磷脂抗体(aPL)具有直接致病性,加之新兴技术为B细胞区“重置”提供了新希望,我们可以推测:更有效地抑制抗磷脂抗体的产生,将有助于改善APS的治疗策略。
研究人员提出了多种基于自身抗原的APS治疗策略,包括能够阻止致病性抗β2GPI抗体与β2GPI结合的药物。同时,目前在全球范围内针对各类自身免疫性疾病正在积极研究的更多策略包括:降低抗体水平的新生儿Fc受体阻滞剂、靶向CD20的抗体(相较于
参考文献

1.Knight JS, Branch DW, Ortel TL. Antiphospholipid syndrome: advances
in diagnosis, pathogenesis, and management. BMJ 2023;380:e069717.
2. Knight JS, Erkan D. Rethinking antiphospholipid syndrome to guide
future management and research. Nat Rev Rheumatol 2024;20(6):377–388.
3.Barbhaiya M, Zuily S, Naden R, et al; ACR/EULAR APS Classification
Criteria Collaborators. The 2023 ACR/
EULAR Antiphospholipid Syndrome Classification Criteria. Arthritis Rheumatol. 2023;75(10):1687–1702.
4. Kmetov ˇ a K, Lonina E, Yalavarthi S, et al. Interaction of the antiphos- pholipid syndrome autoantigen beta-2 glycoprotein I with DNA andneutrophil extracellular traps. Clin Immunol 2023;255:109714.
5.NaveenKumar SK, Tambralli A, Fonseca BM, et al. Low ectonucleoti
dase activity and increased neutrophil-platelet aggregates in patients with antiphospholipid syndrome. Blood 2024;143(12):1193–1197.
6.Meroni PL, Borghi MO. Antiphos-pholipid antibody assays in 2021:look-ing for a predictive value in addition to a diagnostic one. Front immunol .2021;
12:726820.
文案丨
编辑丨王炎焱
医脉通是专业的在线医生平台,“感知世界医学

(本网站所有内容,凡注明来源为“医脉通”,版权均归医脉通所有,未经授权,任何媒体、网站或个人不得转载,否则将追究法律责任,授权转载时须注明“来源:医脉通”。本网注明来源为其他媒体的内容为转载,转载仅作观点分享,版权归原作者所有,如有侵犯版权,请及时联系我们。)








