
帕金森病具有显著异质性,不同患者在前驱表现、临床症状、疾病进展速度和生物学特征方面存在差异。随着遗传学、生物标志物和纵向临床研究不断积累,帕金森病的认识正在逐渐超越单一临床诊断框架。GBA1变异是目前帕金森病最常见的遗传风险因素之一,也与


GBA1基因编码溶酶体酶葡糖脑苷脂酶。该酶在溶酶体内催化葡糖神经酰胺分解为神经酰胺和
GBA1双等位基因致病变异可导致葡糖脑苷脂酶活性不足,使葡糖神经酰胺过度蓄积,并引起
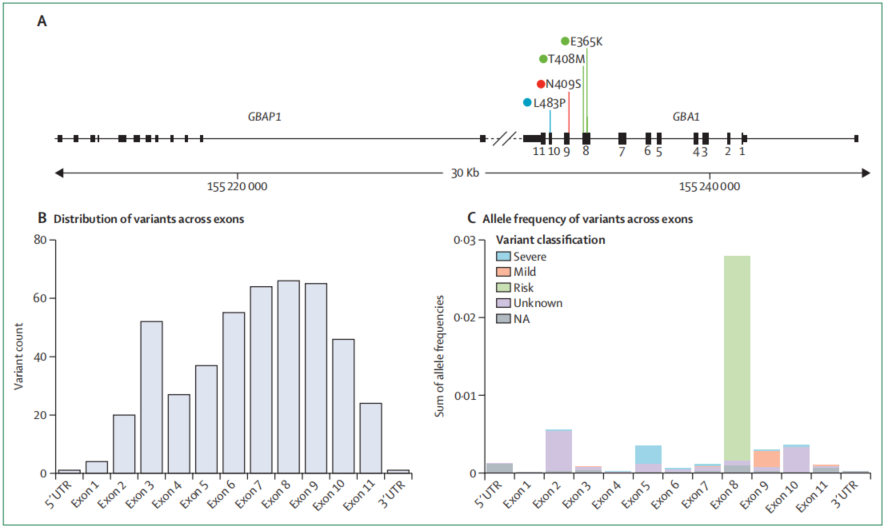
图1 GBA1基因结构及不同类型变异分布
GBA1变异在人群中的分布具有明显差异。欧洲帕金森病患者中,GBA1变异检出比例可达10%–15%;在阿什肯纳兹犹太裔患者中,这一比例可升至20%。不同族群中常见变异并不相同:N409S是犹太人群中最常见的轻度变异,L483P是亚洲和西班牙裔人群中最常见的重度变异,E365K则是欧洲血统人群中最常见的风险变异。近年研究还在非洲和非洲混合血统帕金森病患者中识别出具有族群特征的变异,例如非编码rs3115534-G变异。该变异参与GBA1 mRNA剪接,其存在可导致蛋白表达和酶活性下降。
不同GBA1变异所对应的帕金森病风险并不相同。总体上,重度变异的风险最高,例如L483P的比值比为6.4–30.4,V433L为4.9–6.7;轻度变异次之,例如N409S为2.2–7.8;风险变异相对较低,例如E365K为1.6–5.5。
不过,GBA1变异的外显率较低,并且受到年龄影响。GBA1变异携带者在60岁时发生帕金森病的外显率估计为1.5%–4.7%,80岁时为7.7%–9.1%。多数GBA1杂合变异携带者终生不会发展为帕金森病。同样,大多数戈谢病患者即使携带两个GBA1致病变异,并且葡糖脑苷脂酶活性明显降低,也不会发展为帕金森病。这提示,GBA1变异虽然增加帕金森病风险,但并不能单独决定疾病发生。
目前尚无临床工具能够准确预测GBA1变异携带者是否会转化为帕金森病或路易体痴呆。多个遗传和环境修饰因素仍在研究中。例如,较高的多基因风险评分可独立增加GBA1变异携带者发生帕金森病的风险,携带致病变异且多基因风险评分较高者,帕金森病累积发生率最高。部分研究还提示,农药等特定环境毒物暴露可能与GBA1变异携带者帕金森病风险增加相关。未来仍需进一步完善GBA1变异携带者风险分层,以改进遗传咨询,并为预防性干预研究筛选更合适的人群。

GBA1相关帕金森病的临床表型通常比特发性帕金森病更重,表现为更明显的运动和非运动症状,尤其是认知、情绪、睡眠和自主神经功能受累更突出,疾病总体进展也更快。这些差异在重度变异携带者中最为明显。重度GBA1变异携带者发病年龄较特发性帕金森病患者以及其他类型变异携带者更早,也更容易出现运动并发症,包括运动波动和异动症。
认知受损是GBA1相关帕金森病的重要临床特征。与特发性帕金森病相比,GBA1相关帕金森病患者
非运动症状方面,GBA1相关帕金森病患者情绪障碍更常见,重度变异携带者更为突出。2022年一项Meta分析显示,GBA1相关帕金森病患者快速眼动睡眠行为障碍风险升高,尤其见于L483P重度变异和N409S轻度变异携带者。多项队列研究还报告,与特发性帕金森病相比,GBA1相关帕金森病患者
由于GBA1相关帕金森病患者既容易出现运动并发症,也常伴有复杂非运动症状,器械辅助治疗在这一人群中的应用受到关注。早期回顾性研究汇总12个数据集后显示,接受脑深部电刺激治疗的GBA1变异携带者术后5年内认知下降速度更快,不仅快于接受脑深部电刺激治疗的特发性帕金森病患者,也快于未接受脑深部电刺激治疗的GBA1相关帕金森病患者。在接受脑深部电刺激治疗的GBA1相关帕金森病患者中,轻度或重度变异携带者的认知下降速度快于风险变异携带者。
另一项多中心回顾性研究的5年随访数据显示,GBA1相关帕金森病患者认知功能下降比例为25%,高于特发性帕金森病患者的11%。还有研究提示,GBA1相关帕金森病患者在脑深部电刺激治疗后10年发生认知下降和精神病性事件的风险增加。不过,这些研究同时显示,GBA1相关帕金森病和特发性帕金森病患者接受脑深部电刺激后,运动功能改善和运动并发症减少程度相近。也有较新的研究提出不同结果,认为GBA1相关帕金森病患者无论是否接受脑深部电刺激,发生痴呆的风险均较高。
在

GBA1相关帕金森病的严重表型,尤其是快速认知下降,能否由特定生物化学特征解释,是当前研究的重要问题。近年较受关注的方向是α-突触核蛋白种子扩增检测。在多个大型队列中,与特发性帕金森病相比,GBA1相关帕金森病患者在种子扩增检测中表现出更快的达阈时间和更高的荧光曲线下面积。更快的种子扩增动力学可独立于
这一发现也削弱了阿尔茨海默病共病理在GBA1相关帕金森病认知下降中的解释权重。研究显示,GBA1相关帕金森病和特发性帕金森病患者脑脊液Aβ42和p-tau181等阿尔茨海默病相关标志物水平相似,GBA1相关帕金森病病例中未观察到同时呈现α-突触核蛋白和阿尔茨海默病脑脊液特征的情况,仅有2%的病例呈现孤立阿尔茨海默病特征,且均为N409S轻度变异携带者。死后病理研究也显示,GBA1变异在路易体病理负荷较高、阿尔茨海默病病理负荷较低的病例中更常见。总体来看,当前证据更支持GBA1相关帕金森病的严重临床表型和加速认知下降主要来自α-突触核蛋白病理增强。
相较而言,葡糖脑苷脂酶活性及其底物作为疾病严重程度生物标志物的价值有限。多项研究显示,GBA1相关帕金森病患者干血斑和外周血单个核细胞中葡糖脑苷脂酶活性低于特发性帕金森病患者,且变异越严重,酶活性越低。然而,非常低的酶活性更多提示戈谢病,而绝大多数戈谢病患者并不发展为帕金森病。葡糖神经酰胺、葡糖鞘
葡糖神经酰胺在部分研究中被发现于GBA1相关帕金森病患者血浆和脑脊液中升高,但其与运动或认知功能纵向变化之间的关系并不稳定。有研究显示,脑脊液葡糖神经酰胺与鞘磷脂比值位于最高四分位的GBA1相关帕金森病患者,认知功能下降较最低四分位者更快,但运动功能未见类似关联。不过,临床试验数据显示,即使脑脊液葡糖神经酰胺水平下降,认知功能也未出现改善,这使得葡糖神经酰胺参与认知下降的假设仍需进一步验证。
GBA1相关帕金森病的发病机制主要围绕葡糖脑苷脂酶缺陷、溶酶体功能障碍、线粒体异常、脂质代谢改变和α-突触核蛋白积累展开。不同GBA1变异可能通过不同机制影响葡糖脑苷脂酶,包括功能缺失和功能获得效应。部分变异可导致突变蛋白错误折叠并滞留于内质网,引发内质网应激和相关降解。以L483P为代表的重度变异,以及程度较轻的N409S,可导致葡糖脑苷脂酶在内质网中错误折叠、滞留和降解,并诱导内质网应激;E365K等风险变异则可能涉及不同机制。
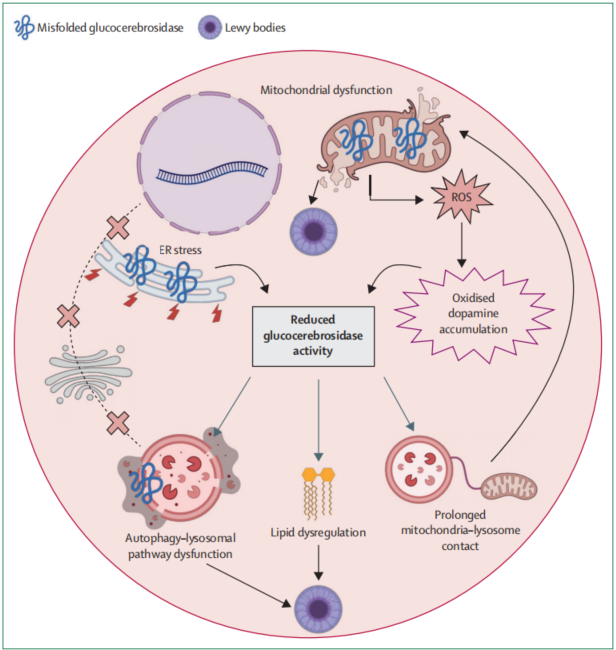
图2 GBA1相关帕金森病涉及的细胞通路
葡糖脑苷脂酶缺陷可破坏自噬-溶酶体功能,并促进α-突触核蛋白积累。研究显示,不同GBA1变异对蛋白稳态影响并不相同。诱导多能干细胞来源
mTOR复合体1也可能参与自噬-溶酶体功能异常。多项GBA1相关帕金森病多巴胺能神经元模型研究发现,葡糖脑苷脂酶缺陷可伴随mTOR复合体1过度激活;在这些模型中,抑制mTOR可逆转α-突触核蛋白聚集并降低内质网应激。线粒体功能异常和氧化应激同样被认为参与GBA1相关帕金森病。研究发现,葡糖脑苷脂酶可进入线粒体,并有助于维持线粒体复合体I完整性和细胞能量代谢。在GBA1变异患者来源的多巴胺能神经元和中脑类器官中,可观察到呼吸链复合体I完整性和细胞能量代谢异常。GBA1变异小鼠脑内还可出现活性氧生成增加和活性氮物质积累,并促进α-突触核蛋白聚集。
脂质代谢改变也参与GBA1相关帕金森病的发病过程。葡糖脑苷脂酶底物葡糖神经酰胺和葡糖鞘氨醇可促进α-突触核蛋白向毒性形式转化。虽然在GBA1相关帕金森病患者死后脑组织中尚缺乏底物累积的稳定证据,但局部溶酶体膜内底物累积仍可能参与发病。脂质膜成分变化还可影响α-突触核蛋白聚集倾向。来自L483P变异GBA1相关帕金森病患者成纤维细胞的研究显示,鞘磷脂、神经酰胺和短链己糖神经酰胺比例升高,长链分子比例下降,短链与长链鞘脂比例升高与葡糖脑苷脂酶活性降低相关;这些患者来源脂质提取物较无神经系统疾病对照来源脂质更能诱导重组人α-突触核蛋白体外纤维化。
α-突触核蛋白在神经元间扩散被认为是帕金森病进展中的重要病理环节。多项细胞和动物模型支持葡糖脑苷脂酶缺陷可促进α-突触核蛋白扩散。葡糖脑苷脂酶缺陷细胞接受预成纤维处理后,鼠皮质神经元中α-突触核蛋白释放增加;携带L483P敲入变异的小鼠在纹状体注射α-突触核蛋白预成纤维后,α-突触核蛋白扩散明显强于野生型同窝小鼠。在另一种延髓迷走神经系统过表达α-突触核蛋白的体内模型中,L483P转基因小鼠α-突触核蛋白由尾侧向头侧扩散加重,可能与线粒体氧化应激有关。不过,目前尚无人体研究直接证明葡糖脑苷脂酶缺陷促进α-突触核蛋白扩散这一关系。

围绕GBA1通路的治疗探索主要包括底物减少治疗、药理伴侣、葡糖脑苷脂酶变构激活剂、基因治疗及其他实验阶段靶点。底物减少治疗的代表药物venglustat可通过抑制葡糖神经酰胺合成酶减少底物累积。MOVES-PD是一项2期、多中心、随机对照试验,纳入221例GBA1相关帕金森病患者。Venglustat显示良好靶点参与,脑脊液和血浆葡糖神经酰胺水平较基线下降约75%,但与安慰剂相比未显示临床获益。该结果使底物减少治疗能否改变GBA1相关帕金森病临床进展仍存在不确定性。
药理伴侣可促进葡糖脑苷脂酶正确折叠并转运至溶酶体,从而提高溶酶体中葡糖脑苷脂酶水平,减少错误折叠蛋白积累及相关细胞损伤。Ambroxol是GBA1相关帕金森病中研究较多的药理伴侣。其作为混合型伴侣分子,在多种体内外模型中显示积极结果。AiM-PD是一项2期开放标签研究,结果显示ambroxol可增加脑脊液中葡糖脑苷脂酶和总α-突触核蛋白水平,并在6个月时改善运动功能。不过,由于该研究为开放标签设计且样本量较小,临床结果仍需谨慎解读。另一项针对轻中度
BIA 28-6156是一种葡糖脑苷脂酶变构激活剂,可在酶活性位点以外结合葡糖脑苷脂酶,防止酶抑制并促进突变蛋白翻译后折叠,从而理论上增加葡糖神经酰胺和葡糖鞘氨醇裂解,并降低溶酶体底物水平。该药已在GBA1相关帕金森病患者中完成1b期研究,连续治疗28天显示安全性、药代动力学和药效学特征良好。不过,研究中在治疗14天时观察到外周血单个核细胞内葡糖神经酰胺短暂升高,随后在28天时回落至给药前水平。尽管该化合物的靶点参与证据尚不清晰,其2期随机对照试验已经启动。
基因治疗旨在恢复GBA1的生理表达及葡糖脑苷脂酶酶活性。当前首个在中重度GBA1相关帕金森病患者中评估脑池内给药基因治疗的人体研究仍处于早期阶段,相关1/2a期研究预计于2029年获得结果。静脉重组葡糖脑苷脂酶已广泛用于戈谢病治疗,安全性良好,但因分子量较大无法穿过血脑屏障。一项1期研究在4例GBA1相关帕金森病患者中评估磁共振引导聚焦超声辅助单侧壳核递送葡糖脑苷脂酶,结果提示该方法安全可行,为大分子蛋白进入脑内提供了一种新的给药方式。
目前,GBA1已经成为GBA1相关帕金森病及部分特发性帕金森病研究中的重要治疗靶点。多项围绕葡糖脑苷脂酶功能恢复、底物代谢调节和基因治疗的临床试验仍在进行中。随着相关研究结果陆续公布,GBA1通路能否成为帕金森病疾病修饰治疗的有效干预方向,还需要更大样本、更长随访的临床研究进一步验证。
参考文献:Menozzi E, Toffoli M, Deleidi M et al.New evidence on the clinical, genetic, and biochemical bases of GBA1-Parkinson's disease: prospects for treatment.The Lancet Neurology, 25, 602-614
 我要投稿
我要投稿















{kind=link}