研读完 MSK‑Sloan 的 5 万例基因检测数据后,再看 EGFR 突变的 NSCLC,尤其是合并 TP53 突变的患者群,我们已经很难再在报告边上只写一句“预后较差”了。MSK‑IMPACT 这类大队列的真正价值,在于把“癌种、驱动基因、克隆性、功能类别”四个要素结合判断:某个突变究竟是支撑肿瘤生长的“主干动力”,还是随波逐流的“伴随乘客”。
聚焦于 EGFR 突变肺腺癌,TP53 几乎交出了一份“全票高危”的答卷:突变率往往超过 50%,多数在原发阶段就已存在;常伴随另一等位基因缺失,形成典型“双等位失活”;从克隆演化来看,它并非疾病晚期才偶然长出的旁支,而是自始至终深植于肿瘤“主干”的核心事件。 换句话说,对相当一部分 EGFR 突变肺癌患者而言,EGFR 突变好比“踩下油门”,而 TP53 突变则相当于“拆掉刹车”,两者共同决定了肿瘤启动与狂飙的方式。这种“油门与刹车双重失控”的底层基因架构,正是我们在临床实践中反复遭遇其难治与早耐药的源头。
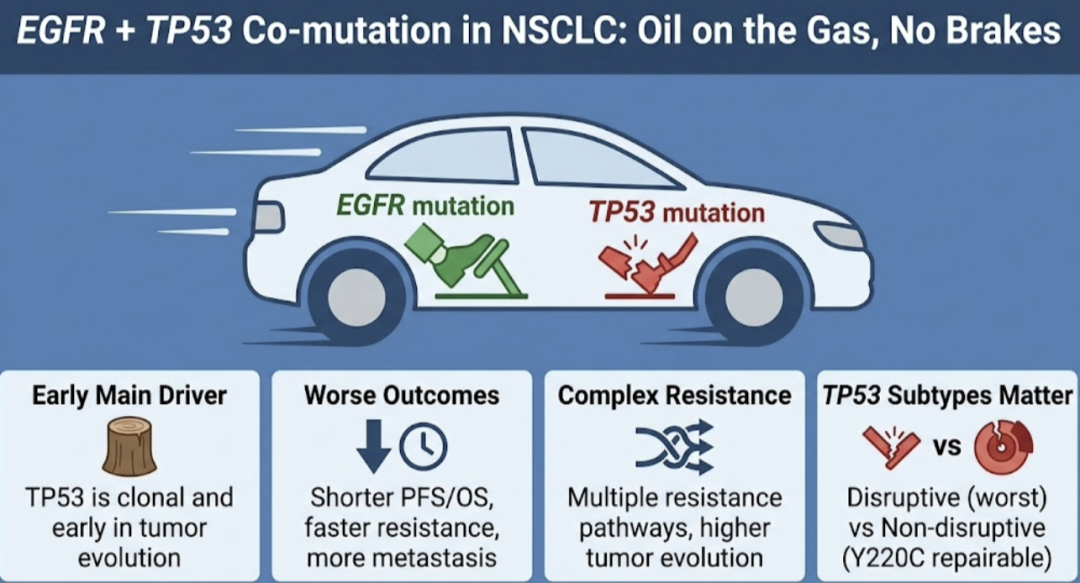
这一点在历代 EGFR‑TKI 的随访数据中屡被印证。第一代 TKI 时代,无论欧美试验还是亚洲真实世界,只要按 TP53 野生/突变分层,EGFR/TP53 共突变患者在客观缓解率(ORR)、无进展生存期(PFS)和总生存期(OS)上几乎“全线落败”,中位 OS 缩短十余个月的现象并不少见。 进入
更棘手的是耐药模式。以往我们常用单一机制来描述 EGFR‑TKI 失败——一代 TKI 后的 T790M,三代 TKI 后的 C797S 或 MET 扩增。但在 EGFR+TP53 共突变人群中,进展时看到的往往是一幅“错综复杂”的耐药图谱:C797S、MET/HER2 扩增、KRAS/BRAF 激活、结构重排,甚至向
对临床来说,理解 TP53 亚型,可以先把它想象成“刹车片坏掉的不同方式”。同样是这块刹车,有的直接断成两半(截断型 / disruptive),几乎完全失灵;有的只是磨得很薄、打滑明显(一般 missense / non‑disruptive),还有少数是缺了一块刚好形成“小坑”,虽然不合格,却给将来用特殊零件“垫一块”修补留了空间(如 Y220C 这类可药物化热点)。
正如 ELCC 2026 上 TOP 研究引发的广泛讨论,TP53 的故事远不止“有无突变”这么简单。即便只看 EGFR 突变 NSCLC,换到分子语言里,上述“刹车坏法”对应的,就是不同类型、不同结构域、不同功能后果的 TP53 突变,而 Poeta 提出的 disruptive/non‑disruptive 分型,正是目前比较好理解的一种分类方法。
基于 Poeta 功能分型,TP53 大致可分为 disruptive 与 non‑disruptive 两大类。前者包括各种提前终止信号(无义、移码、致截断剪接)以及位于 L2/L3 环(163–195、236–251)的特定错义突变,意味着蛋白在结构与功能层面遭遇“毁灭性破坏”;后者则多为保留基本骨架、仅部分丧失功能的错义变异。 多个 EGFR‑NSCLC 队列一再表明:disruptive 变异,尤其是集中在 DNA‑binding 核心区、伴双等位失活者,几乎总是与最短的 PFS/OS 以及最高的小细胞转化风险深度绑定;而部分外围错义突变,其预后则处于完全野生与高度破坏之间的“中间地带”。 若进一步叠加 RB1 等“谱系调控基因”的突变,EGFR+TP53+RB1 这一亚群发生小细胞样转化的概率可达普通 EGFRm 人群的数倍,是名副其实的“极高危”分子亚型。
从严谨性出发,目前的 disruptive/non‑disruptive 更适合作为揭示 TP53 内部功能异质性的研究目标,但现阶段的随机研究(如 FLAURA2、TOP)公开的仍是“TP53 突变整体亚组”的疗效数据,尚缺乏按功能亚型分层的 Kaplan–Meier 曲线与预设交互检验。 更务实的做法,是在病历和 MDT 中尽可能详尽标注 TP53 的位点与类型,把这些信息视为未来前瞻研究和真实世界分析的重要素材。
MSK‑50K 在 driver‑neoantigen 与 HLA‑LOH 层面的分析,为 EGFR+TP53 的免疫学特征提供了一个很好的参照框架。 多个队列显示,一旦合并 TP53,EGFR 突变肿瘤整体 PD‑L1 表达和 TMB 水平明显上升,免疫微环境更接近“热肿瘤”,这与临床上少数 TKI 失败后对免疫联合化疗有反应的 EGFR 患者相吻合。 用 MSK‑50K 的话说,TP53 失活在 driver‑neoantigen 层面给肿瘤贴上了更多“通缉犯”的标签。
但更高的免疫原性同样伴随更高的逃逸潜力。MSK‑50K 指出,当 driver‑neoantigen 与特定 HLA 配型高度匹配时,肿瘤可以通过选择性丢失该等位基因(HLA‑LOH)来“拆掉告示牌”,从而在免疫监视之下实现隐身。 对 EGFR+TP53 来说,这意味着:一方面,它可能是 EGFRm 人群中最有机会从 TCR、双特异抗体等精细免疫策略中获益的亚群;另一方面,也可能是最需要动态监测 HLA‑LOH、最容易在免疫压力下快速重塑抗原谱的那一群。如何在“免疫可及”与“免疫逃逸”之间找到真正的治疗窗口,本身就值得专门设计研究。
如果前面的焦点都在于“TP53 有多坏”,那么围绕 Y220C 这类可药物化热点的进展,则把话题推进到另一个层面:有没有机会在体内直接“修好”一部分 p53?
在高级别浆液性卵巢癌中,TP53 几乎普遍突变,其中 Y220C 是相对集中的热点之一,因此被选作 p53 结构再激活药物 rezatapopt(PC14586)等的首批适应证。 现有Ⅰ/Ⅱ期数据提示,在携带 Y220C 的卵巢癌及部分其他实体瘤中,rezatapopt 确实可以获得一定的客观缓解和疾病控制,证明“用小分子嵌入突变口袋、稳定畸形 p53 构象、部分恢复功能”在人体内并非天方夜谭。
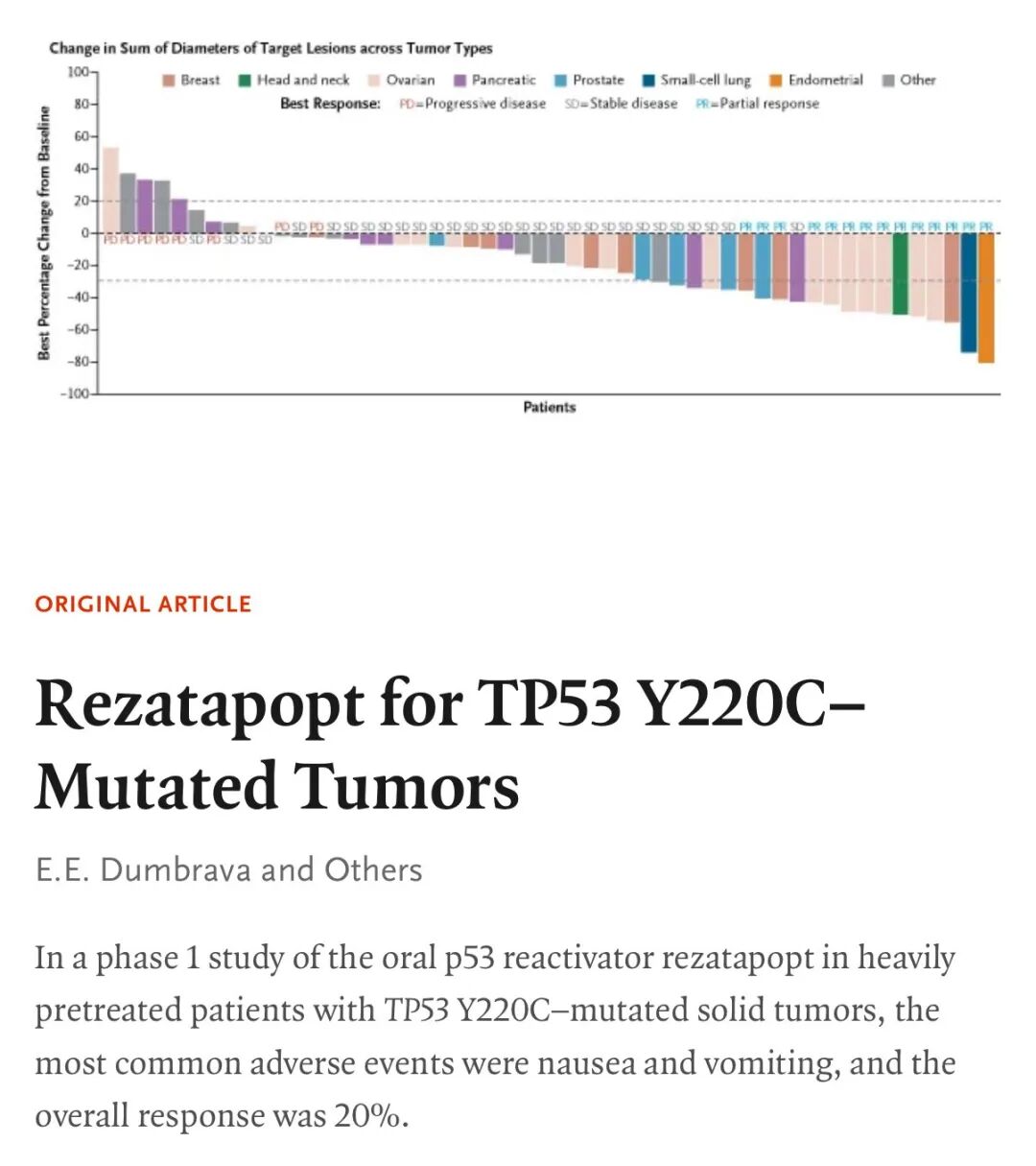
从结构上看,Y220C 落在 TP53 的 DNA‑binding domain 内,但不在 Poeta 定义的 L2/L3 环,因此被归入 non‑disruptive 一类。 Tyr→Cys 的“大到小”替换在局部形成一个疏水空腔,使蛋白热不稳定、功能部分丧失,却没有让整体折叠彻底崩溃。 正是这点,让 Y220C 既“足够致病”,又“仍可扶正”:一方面明显削弱了 p53 抑癌功能,另一方面又为小分子提供了一个稳定的嵌入位点。
对 NSCLC 而言,这条路在生物学上是打开的——只要肿瘤携带 TP53‑Y220C,理论上都存在被 p53 再激活小分子“修补”的可能。目前的 PC14586 篮子试验也确实纳入了少量肺癌病例,但迄今披露的临床数据几乎全部来自卵巢癌等 TP53 高负荷肿瘤,尚无专门针对 NSCLC 的成熟结果,更谈不上改变 EGFR+TP53 人群一线治疗格局。
短期内,EGFR+TP53 NSCLC 的主战场仍然是基于 EGFR‑TKI 的一线强化(如 FLAURA2、TOP 模式)以及后线联合,而不是指望 p53 靶向药“取代”这些策略。中长期看,针对少数可药物化位点(以 Y220C 为代表)的 p53 再激活药,确实有机会为一小部分 EGFR+TP53 患者打开新的精准治疗通路——前提是,我们今天在基因报告和 MDT 中,已经开始认真记录“TP53 到底是哪一型”,为未来试验和联合预留空间。
审核:Faline
排版:Faline
执行:Faline
(本网站所有内容,凡注明来源为“医脉通”,版权均归医脉通所有,未经授权,任何媒体、网站或个人不得转载,否则将追究法律责任,授权转载时须注明“来源:医脉通”。本网注明来源为其他媒体的内容为转载,转载仅作观点分享,版权归原作者所有,如有侵犯版权,请及时联系我们。)








