近期,《Frontiers in Medicine》期刊发表了一篇病例:“Alport syndrome complicated with IgA nephropathy: a case report”,该病例通过肾活检与基因检测明确共病诊断,为临床精准诊疗提供实证。
患者女性,29岁,因发现蛋白尿于2023年3月11日入院。
孕13周产检时首次发现蛋白尿,孕期多次检查尿蛋白均持续为3+。患者于
父母体健,无肾脏疾病家族史。
入院时血压122/72 mmHg,双下肢无
免疫荧光检查示:IgA(+++)、IgM(+)、C3(++)、肾小管重吸收颗粒(+),其余标志物阴性。
光镜下可见15个肾小球,其中1个节段性肾小球硬化(图1A、1B);肾小球系膜细胞及基质轻度增生,系膜区可见红细胞沉积,毛细血管袢开放(图1C)。肾小管上皮细胞可见空泡变性及颗粒变性,局灶肾小管萎缩,伴间质炎性细胞浸润(无纤维化)。肾小球毛细血管内皮可见空泡变性,肾小囊上皮细胞空泡变性,无增生。肾小球基底膜(GBM) 厚度不一,约140~600 nm,致密层增厚;部分区域可见撕裂及蛛网状改变(图1D)。上皮细胞肿胀、空泡变性,足突广泛融合,可见系膜细胞及基质增生。
电镜下见系膜区电子致密物沉积,肾小管间质区域内肾小管上皮细胞空泡变性。毛细血管管腔内可见红细胞,间质见泡沫细胞,肾间质无特征性病变。观察到肾小球基底膜撕裂,高度提示Alport 综合征。
本例病理符合轻度系膜增生性IgA肾病,按Lee分级为Ⅱ级,按IgA肾病牛津分型为M1 E0 S1 T0 C0。
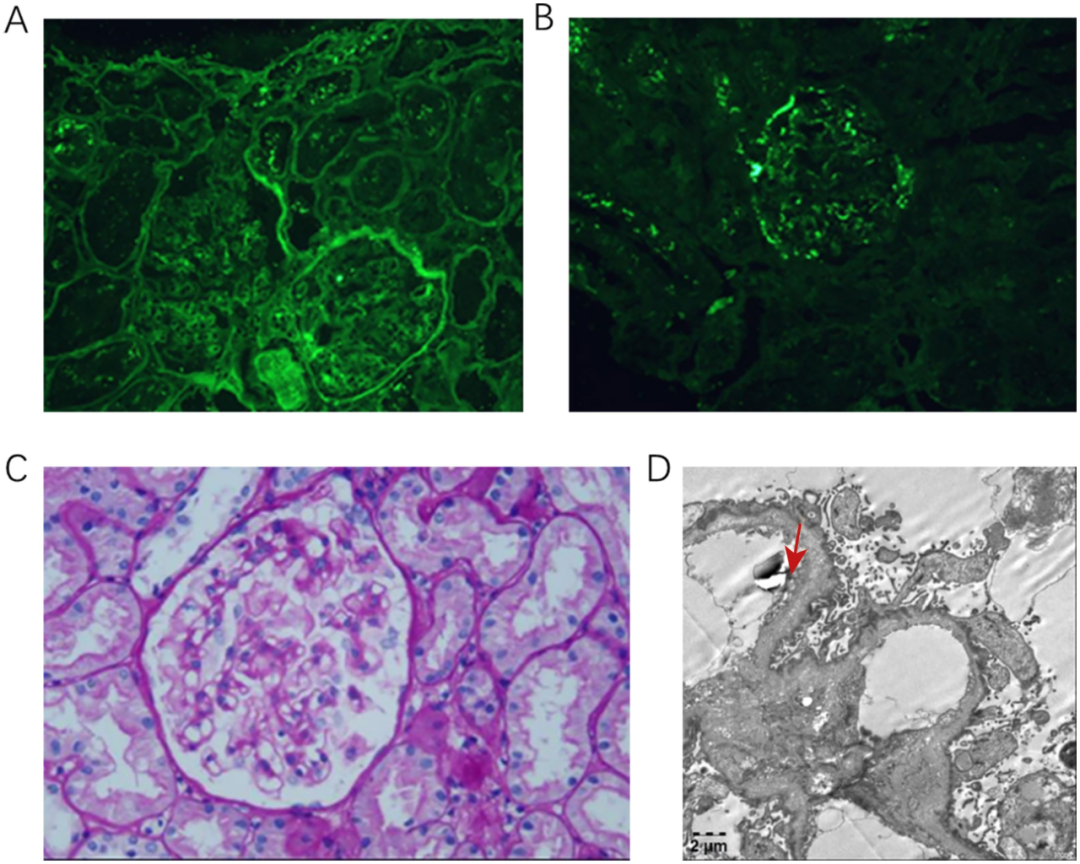
图 肾脏
(A、B)IgA呈颗粒状沉积于系膜区。
(C)光镜所见:肾小球系膜细胞及基质轻度增生。
(D)电镜所见:毛细血管基底膜厚薄不均,厚度约为140–600 nm。基底膜致密层增厚,部分区域呈撕裂状及蜘蛛网样改变。
患者病程中持续接受血管紧张素Ⅱ受体拮抗剂(ARB)联合钠
结合上述结果,最终诊断:Alport综合征合并IgA肾病。继续予吗替麦考酚酯、醋酸泼尼松、SGLT2i 及
本例患者同时罹患IgA肾病与Alport综合征,临床表现为血尿、蛋白尿及肾功能损害,但二者病因与病理机制截然不同。该病例强调了结合组织病理学、电镜检查与基因检测实现精准诊断的重要性。同时需指出,本例患者受经济条件限制,未行Ⅳ型胶原免疫荧光检测。
遗传性IgA肾病作为IgA肾病的亚型之一,遗传方式包括X连锁隐性遗传及常
Alport 综合征与IgA肾病均可表现为血尿和蛋白尿,临床鉴别存在一定难度。有报道指出,部分初诊为IgA肾病的患者,经基因或超微结构分析后最终确诊为Alport综合征,反之亦然。此外,两种疾病亦可共存于同一家系或同一患者体内,进一步增加诊断难度,并可能影响治疗策略的选择。在病理机制上,Alport综合征患者因基因缺陷导致肾小球基底膜结构薄弱,更易受IgA肾病免疫复合物沉积的继发性损伤。基底膜基质异常可促进免疫复合物异常沉积与持续滞留,加剧炎症反应,进而协同加速肾小球损伤进程。
疾病共存增加了临床管理的复杂性。以IgA肾病为例,常用免疫抑制治疗,但对于以结构缺陷为主而非炎症活动的Alport综合征患者,该疗法可能疗效欠佳甚至风险更高。因此,对此类病例必须采取个体化策略,并通过基因检测及电镜检查进行精准诊断。
本例患者因初诊时未行基因检测,初始治疗主要针对IgA肾病,方案包括ARB、SGLT2抑制剂、霉酚酸酯(MMF)及小剂量糖皮质激素。尽管ACEI/ARB是两者的基础治疗,但单用疗效有限,且因高钾风险而禁忌联用。新兴药物Sparsentan(双重内皮素-血管紧张素受体拮抗剂)在降低蛋白尿、延缓肾损伤方面显示出潜力,且可改善听力,目前已在美国获批用于IgA肾病,但在Alport综合征中的疗效尚待验证。针对IgA肾病,国内大型随机对照试验证实MMF联合小剂量激素有效且安全,因此该方案在国内临床广泛应用,但循证依据支持其作为激素节省剂,且主要适用于中国患者。基于此,本例亦采用该方案。此外,羟氯喹通过抑制Toll样受体与细胞因子发挥免疫调节作用,兼具减少两种疾病蛋白尿的潜力,但目前多数疗法尚处研究阶段。分子伴侣、干细胞及基因治疗等已进入临床试验,创新疗法的迅速发展有望为患者带来新希望。
本病例强调了肾活检和基因检测在实现精确诊断中的关键作用,二者对于及时启动适当治疗至关重要。该合并Alport综合征和IgA肾病的患者,在接受霉酚酸酯、醋酸泼尼松和厄贝沙坦方案治疗后,未能获得显著的临床获益,这凸显了开展针对性诊断和制定个体化治疗策略的必要性。
参考文献:
Sun J and Yu F (2026) Alport syndrome complicated with IgA nephropathy: a case report. Front. Med. 13:1739845. doi: 10.3389/fmed.2026.1739845
医脉通是专业的在线医生平台,“感知世界医学脉搏,助力中国临床决策”是平台的使命。医脉通旗下拥有「临床指南」「用药参考」「医学文献王」「医知源」「e研通」「e脉播」等系列产品,全面满足医学工作者临床决策、获取新知及提升科研效率等方面的需求。仅供HCP观看。
 我要投稿
我要投稿















{kind=link}