导读
本研究采用了一套整合多组学与生物工程技术的系统性研究策略,旨在深入剖析基质刚度对系统性硬化症(SSc)成纤维细胞行为与命运的调控作用。首先,构建了一个基于Ⅰ型胶原的三维(3D)细胞培养模型,通过精确控制聚合温度(16℃、27℃、37℃)来生成具有梯度刚度的基质,以此模拟体内纤维化进程中细胞外基质(ECM)力学特性的动态变化。随后,利用该模型,对来源于SSc患者及健康对照的皮肤成纤维细胞进行了批量RNA测序(bulk RNA-seq),以识别由基质刚度变化所引发的全基因组转录响应,并从中定义出一组“刚度响应性基因特征”。为将这些体外发现的力学特征与体内疾病状态相关联,研究整合了多个已发表的SSc皮肤单细胞RNA测序(scRNA-seq)数据集,将上述刚度特征映射到单细胞分辨率,从而在复杂的成纤维细胞异质性群体中鉴定出对力学信号最为敏感的特定细胞亚群。进一步,对scRNA-seq数据应用轨迹推断算法(如PHATE和Slingshot),以构建这些刚度敏感细胞的分化路径。为验证力学传导的核心机制,分析了在机械应变条件下施用黏着斑激酶(FAK)抑制剂的成纤维细胞scRNA-seq数据。最后,利用空间转录组学技术,并借助Cytospace工具将单细胞簇对齐至组织空间位置,以在完整的组织架构中可视化关键成纤维细胞亚群的分布,并评估其定位与局部ECM刚度及沉积程度的空间关联。这一系列方法构成了一个从体外机制探索到体内验证的完整研究闭环。
通过调节聚合温度,成功构建出具有显著差异纤维直径与孔隙度的胶原基质(图1)。纤维束厚度随温度降低而增加,这与前期研究中刚度升高的趋势一致。细胞活力在不同密度下保持稳定,且纤维结构特征与小鼠体内皮肤ECM相似,支持该模型的生理相关性(图1)。

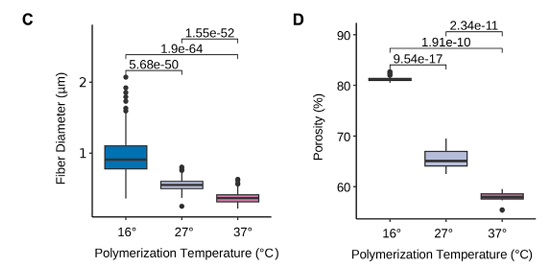
图1:胶原基质中聚合温度依赖的微观结构变化:多模式成像与定量分析
批量RNA-seq分析显示,无论是SSc还是正常成纤维细胞,在高刚度基质中均出现显著的转录组重编程(图2)。上调基因富集于纤维化相关通路与力学传导通路,涉及RACK1、LGALS1等关键分子,表明成纤维细胞能敏锐感知并响应力学微环境变化。
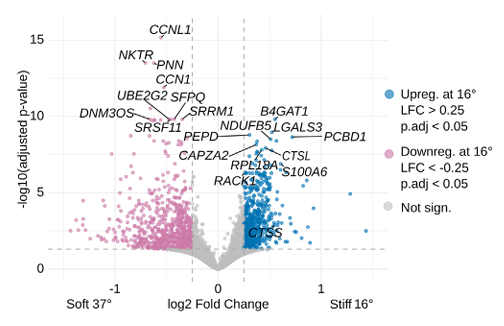
图2:16℃和37℃细胞之间差异基因表达(DGE)的火山图,已对协变量进行调整
整合scRNA-seq数据发现,SSc皮肤成纤维细胞可分为SFRP2⁺,CXCL12⁺,TNN⁺和CLDN1⁺等主要亚群(图3C)。其中,SFRP2⁺成纤维细胞中PI16⁺亚群对刚度响应最为显著(图3D),且在SSc中其响应性进一步升高(图3D右)。该亚群同时高表达机械敏感离子通道PIEZO1与PIEZO2(图3E),提示其具备较强的力学感知能力。
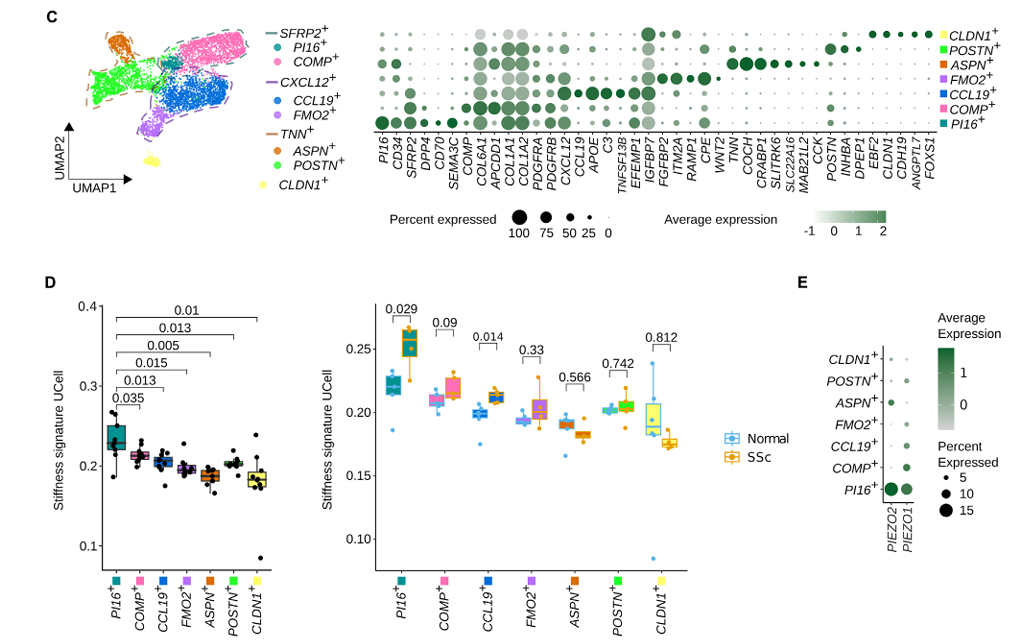
图3:PI16+成纤维细胞在系统性硬化症的成纤维细胞亚群中对基质刚度的反应最强
转录相似性与层次聚类分析显示,PI16⁺细胞与同属SFRP2⁺区域的COMP⁺成纤维细胞具有高度相似性(图4A)。SSc患者中PI16⁺/COMP⁺比率显著降低(图4B),提示PI16⁺向COMP⁺分化活跃。PHATE轨迹分析进一步揭示一条从PI16⁺向COMP⁺发展的连续分化路径(图4C),沿该轨迹TGF-β与IL-4刺激的促纤维化信号评分逐步升高(图4D)。COMP⁺细胞在SSc中上调COL1A1、COL1A2等ECM合成基因,并激活ECM重塑与粘附相关通路(图4E-F)。
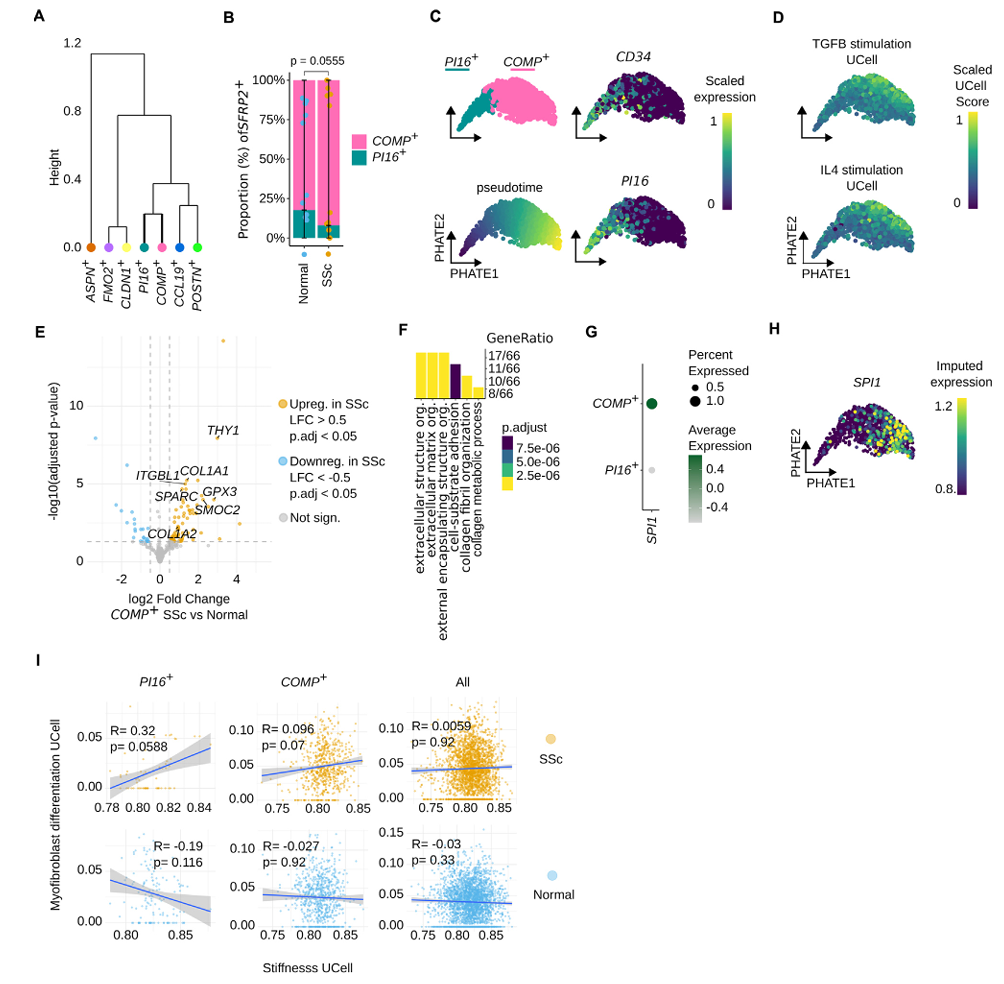
图4:基质刚度增加会诱导PI16+成纤维细胞向肌成纤维细胞转分化
COMP⁺细胞中转录因子SPI1(PU.1)表达高于PI16⁺细胞(图4G),经ALRA插补后信号进一步明确(图4H)。在SSc来源的PI16⁺细胞中,刚度评分与肌成纤维细胞分化评分(GO:0036446)呈显著正相关(图4I),提示刚度驱动其向肌成纤维细胞分化。
通路评分显示Hippo通路及其效应分子YAP1在SFRP2⁺成纤维细胞中显著上调(图5A-B)。FAK(PTK2)在SSc的PI16⁺细胞中显著表达升高(图5C)。在伤口愈合scRNA-seq数据集(GSE167339)中,机械应变可诱导刚度响应特征及COMP⁺特征,而FAK抑制可逆转该效应(图5D-F),证实力学传导是PI16⁺向COMP⁺分化的关键驱动机制。
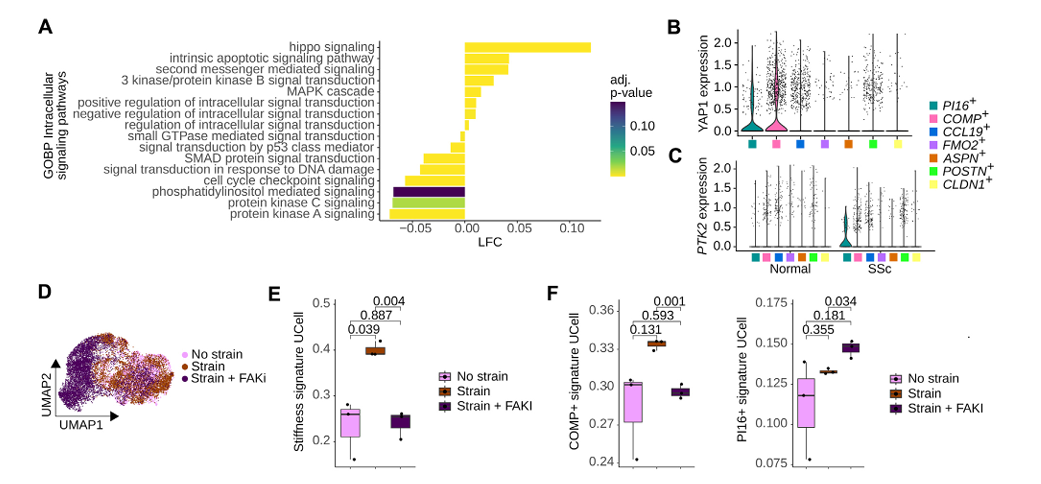
图5:抑制机械转导途径可阻断由刚度引起的肌成纤维细胞活化
空间转录组分析显示,PI16⁺与COMP⁺成纤维细胞在组织内空间共定位(图6A-C),且共定位区域刚度评分与ECM评分均显著升高(图6D-E)。晚期SSc患者皮肤中刚度特征基因表达显著高于早期患者(图6F),进一步支持刚度随纤维化进展而增加。
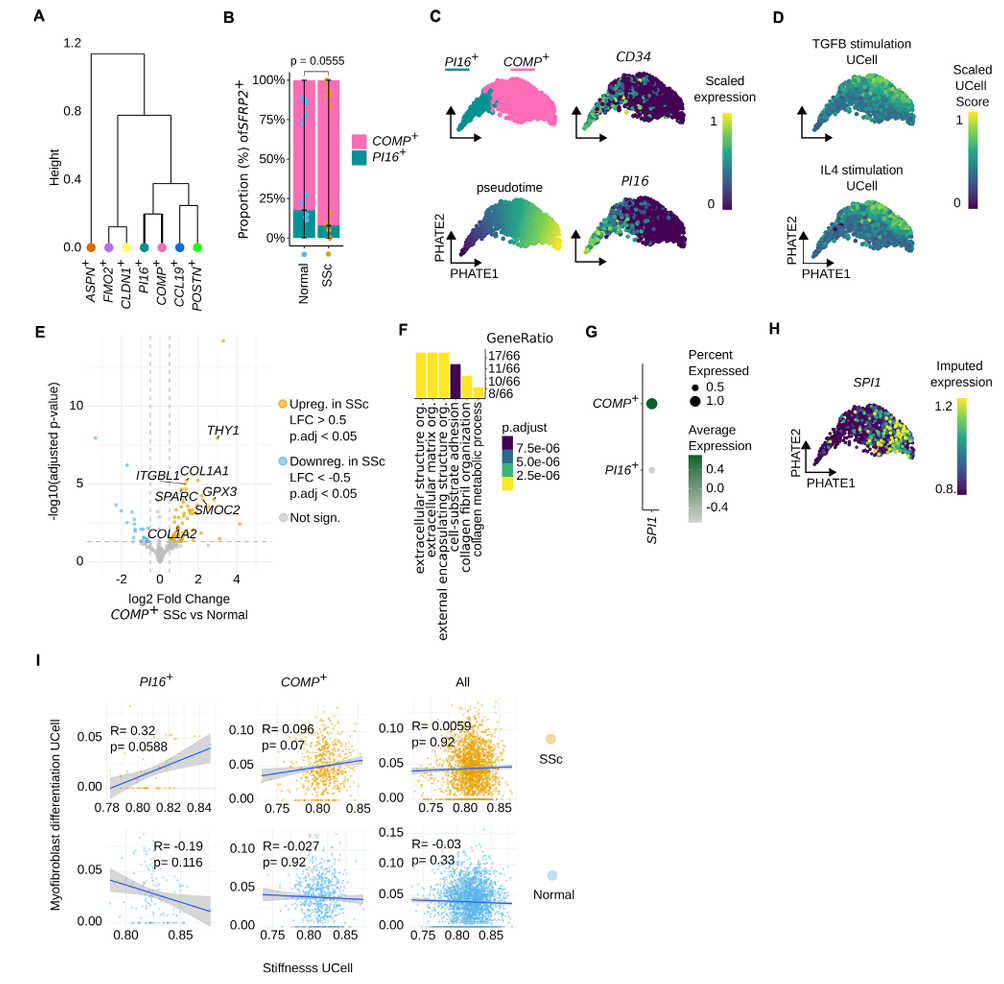
图6:PI16+和COMP+成纤维细胞在纤维化微环境中共定位,该微环境以基质刚度增加为特征
从治疗角度看,靶向ECM刚度或相关力学传导通路(如FAK、PIEZO通道、YAP/TAZ信号)可能为干预纤维化提供新途径。例如,开发小分子抑制剂或抗体以阻断刚度感应、力学信号下传,或靶向特定成纤维细胞亚群,更全面覆盖原文提及的治疗方向,或通过纳米材料调节局部基质力学性能,均为值得探索的方向。
综上,本研究确立了基质刚度作为SSc纤维化核心驱动因素的地位,并为开发基于力学微环境调控的抗纤维化疗法提供了理论依据与潜在靶点。未来研究应进一步阐明刚度感知与信号转导的具体分子机制,并探索其在其他纤维化疾病中的普适性。
参考文献:Ueberall L, Mohammadian H, Demmler R, Ariza Y, Tripal P, Anchang CG, Weber S, Angeli MR, Raimondo MG, Chang J, Huang K, Distler JHW, Distler O, Rauber S, Schett G, Ramming A, Ramming AM. Matrix stiffness regulates profibrotic fibroblast differentiation and fibrotic niche activation in systemic sclerosis. Ann Rheum Dis. 2025 Nov;84(11):1865-1876. doi: 10.1016/j.ard.2025.05.016. Epub 2025 Jun 26. PMID: 40579317.
医脉通是专业的在线医生平台,“感知世界医学脉搏,助力中国临床决策”是平台的使命。医脉通旗下拥有「临床指南」「用药参考」「医学文献王」「医知源」「e研通」「e脉播」等系列产品,全面满足医学工作者临床决策、获取新知及提升科研效率等方面的需求。

(本网站所有内容,凡注明来源为“医脉通”,版权均归医脉通所有,未经授权,任何媒体、网站或个人不得转载,否则将追究法律责任,授权转载时须注明“来源:医脉通”。本网注明来源为其他媒体的内容为转载,转载仅作观点分享,版权归原作者所有,如有侵犯版权,请及时联系我们。)








