编者按
白癜风是典型的CD8+ T细胞介导自身免疫病,具有高度黑色素细胞靶向性,患者及小鼠模型中,分泌IFNγ的黑色素细胞抗原特异性CD8+ T细胞是破坏黑色素细胞的主要效应细胞,且可分化为功能性记忆T细胞。先天免疫细胞、树突状细胞、角质形成细胞及成纤维细胞的协同作用,共同驱动针对黑色素细胞的自身免疫病理过程。过去二十年,通过患者组织样本分析与小鼠模型机制研究,白癜风的免疫触发、耐受打破及疾病持续机制已被系统揭示,相关研究也深化了对CD8+ T细胞自身抗原记忆及组织驻留记忆的认知。本文聚焦白癜风免疫学核心问题,阐述黑色素细胞破坏机制、异常T细胞应答特征及临床干预策略。


1.1 遗传与环境因素


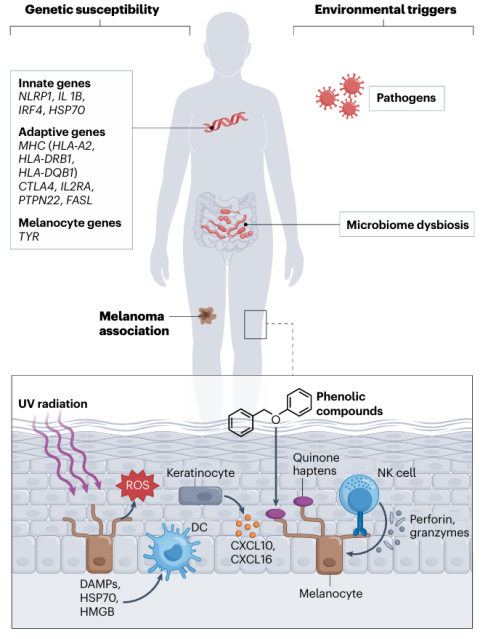


3.1 IFNγ:疾病发生的核心细胞因子
白癜风是典型的IFNγ依赖性疾病,进展期患者皮肤存在特异性IFNγ信号特征,CD8+ T细胞是IFNγ的主要产生者。IFNγ可通过诱导黑色素细胞从表皮基底层迁移失稳,或通过CXCR3B介导凋亡直接杀伤黑色素细胞。
小鼠模型中,IFNγ受体敲除小鼠不发生白癜风,中和IFNγ可预防疾病发生;JAK1抑制剂可阻断IFNγ受体信号,抑制CD8+ T细胞皮肤浸润并预防脱色,靶向表皮的IFNγ双特异性抗体可诱导局部复色,明确了IFNγ作为治疗靶点的核心价值。
3.2 CXCR3-CXCL9/10/11轴:CD8+ T细胞的募集与定位
IFNγ可诱导角质形成细胞、成纤维细胞分泌CXCL9、CXCL10、CXCL11,通过CXCR3募集CD8+ T细胞。Cxcr3敲除小鼠的gp100特异性T细胞无法诱导白癜风,抗CXCR3抗体可逆转疾病并诱导毛囊周围复色。
CXCL9主要促进CD8+ T细胞皮肤募集,而CXCL10是T细胞表皮定位、活化及疾病发生的必需因子,二者在患者皮损中均呈高表达,是介导CD8+ T细胞浸润的关键通路。
3.3 角质形成细胞与成纤维细胞:非T细胞的关键调控作用
皮损中T细胞对IFNγ的应答微弱,角质形成细胞和成纤维细胞的旁分泌应答是疾病发生的必要条件。角质形成细胞特异性缺失IFNγ应答的小鼠,CD8+ T细胞募集及白癜风显著减轻,其产生的CXCL9、CXCL10是T细胞表皮浸润的主要来源;成纤维细胞是患者皮肤中IFNγ信号评分最高的细胞,其特异性敲除IFNγ受体可完全阻止小鼠白癜风发生,提示与角质形成细胞存在功能冗余。
皮肤不同区域成纤维细胞产生趋化因子的能力存在差异,或可解释非节段型白癜风的双侧对称分布;创伤愈合中成纤维细胞的活化可能触发皮损同形反应(图3)。
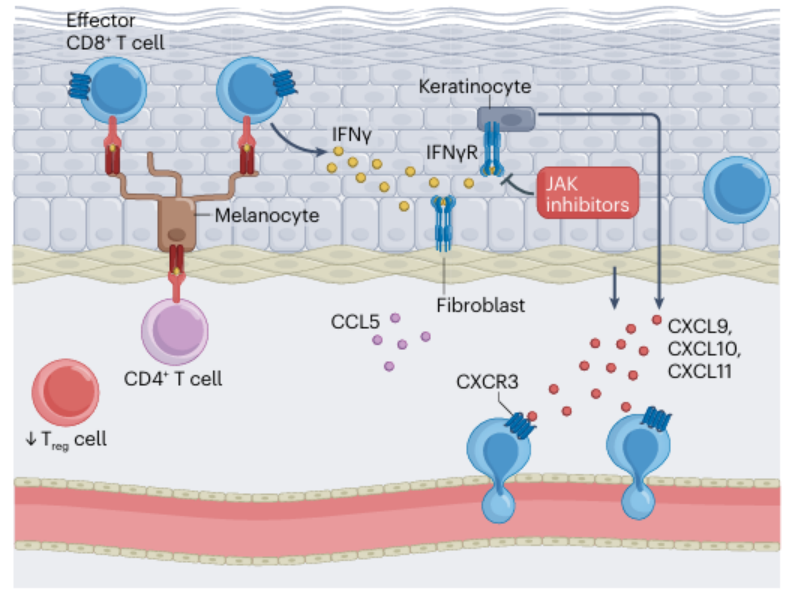
图3:IFNγ、角质形成细胞、成纤维细胞和CD8+T细胞在白癜风中的关键作用
3.4 IL-17与TNF:非疾病驱动性细胞因子
白癜风皮损CD8+ T细胞仅分泌IFNγ而不产生IL-17,与银屑病形成鲜明对比。TNF在皮损中水平升高,但抗TNF治疗效果微弱,且长期使用可增加白癜风发病风险,推测其可能通过打破细胞因子稳态诱发疾病,并非白癜风的核心调控因子。


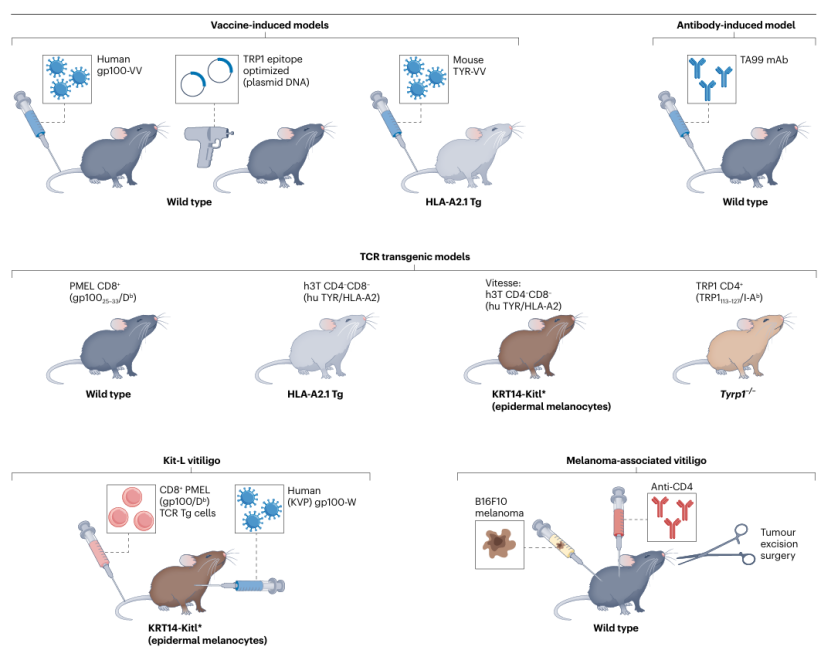
白癜风患者皮肤中MDA特异性CD8+ T细胞呈TRM表型(CD103+CD69+CD49a+),且在皮损及非皮损区均有分布,提示全皮肤记忆细胞定植;MAV患者皮肤存在高表达IFNG或TOX的TRM亚群,小鼠模型中TRM可遍布全皮肤并存在于引流淋巴结,介导长期抗黑色素瘤抵抗。
TRM的维持依赖皮肤局部微环境,其定位于毛囊附近淋巴样聚集体,通过CXCR6与DC表面的CXCL16结合维持存活,毛囊角质形成细胞分泌的IL-15、IL-7是TRM维持的关键细胞因子,且白发毛囊中TRM数量多于黑发毛囊,提示抗原清除可能促进TRM分化。
4.2 循环记忆与TRM的功能分工
CD8+ T细胞记忆在白癜风中持续存在且无功能耗竭,MAV患者的T细胞克隆型可存续长达9年,小鼠模型中针对MDA的CD8+ T细胞可长期分泌IFNγ。IL-15是记忆T细胞维持的关键,中和IL-15可清除TRM并诱导复色,长期治疗可同时清除循环记忆T细胞,实现疾病持久逆转。
循环记忆T细胞与TRM具有明确功能分工:循环记忆T细胞是疾病持续的必需因素,阻断其再循环可逆转白癜风;TRM则主要介导疾病播散与复发,CD103或CXCR6敲除可阻止白癜风超越原始皮损扩散,且JAK抑制剂治疗后疾病在原部位复发,推测TRM通过“感知-警报”功能募集循环记忆T细胞,其自身分泌的CXCL9、CXCL10进一步促进CD8+ T细胞表皮浸润(图4)。
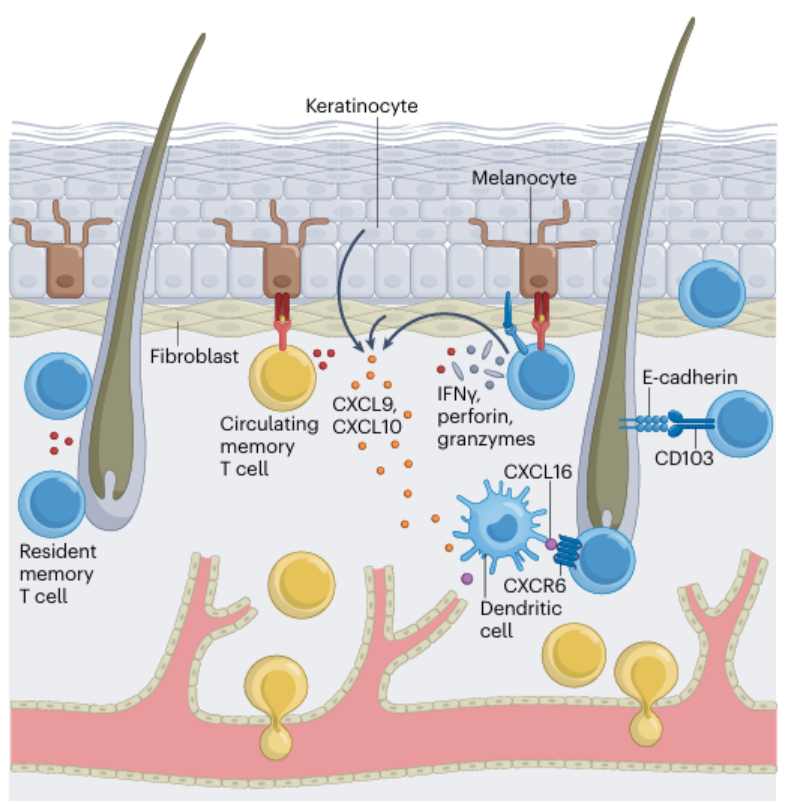
图4:记忆性CD8+T细胞在白癜风中的关键作用


5.1 促进黑色素细胞再生与复色
促复色策略的效果依赖毛囊内残留的黑色素细胞,窄谱UVB是临床一线疗法,其可通过诱导α-MSH释放,结合MC1R刺激黑色素细胞生成黑色素;α-MSH类似物阿法诺肽联合UVB光疗,可更早、更高比例促进复色,疗效优于单药。
Wnt/β-catenin通路激动剂可诱导人皮肤外植体复色,是潜在的促复色靶点;对于完全脱色患者,黑色素细胞移植、激光消融联合皮肤细胞喷雾移植等外科手段可实现持久复色,相关微创疗法正处于临床试验阶段。
5.2 靶向抑制CD8+ T细胞的活化与募集
MDA特异性CD8+ T细胞是免疫抑制治疗的核心靶点,钙调神经磷酸酶抑制剂局部应用可特异性抑制T细胞活化,是活动期皮损的常用药物;JAK抑制剂是目前最具前景的靶向药物,近日,外用JAK抑制剂磷酸
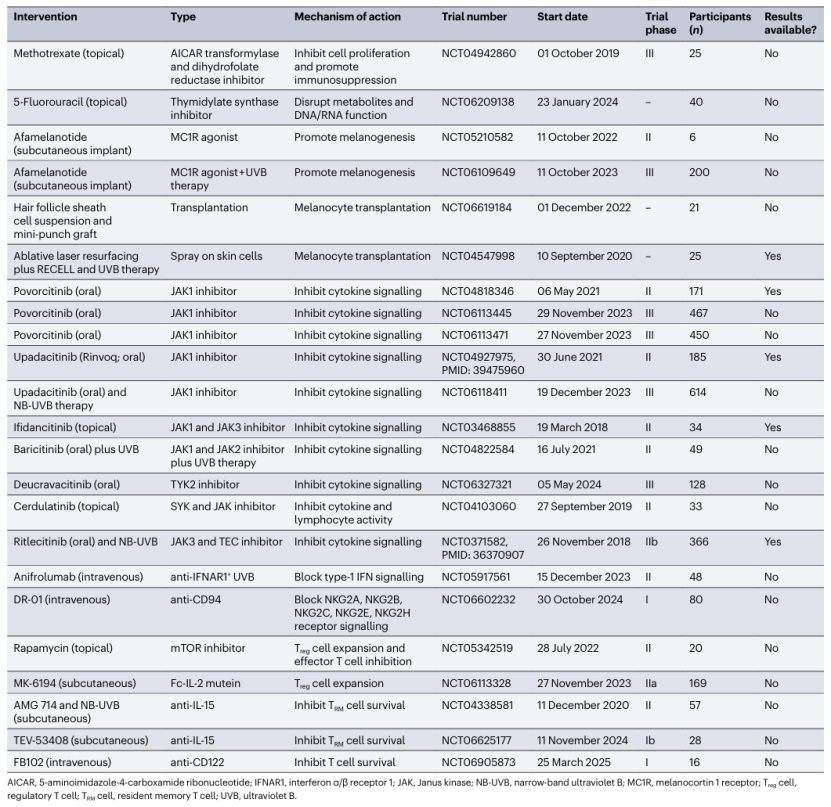
其他JAK家族抑制剂(如JAK1/JAK3、JAK3/TEC抑制剂)单用或联合光疗的临床试验正在进行;针对NKG2D结合伴侣CD94的抗体,可阻断CD8+ T细胞对黑色素细胞的靶向杀伤,是潜在的新型治疗手段(详见表1)。
表1:正在进行和近期针对白癜风患者的临床试验
5.3 增强Treg细胞的外周耐受功能
增强Treg介导的免疫抑制是白癜风的重要治疗方向,雷帕霉素可通过抑制mTOR通路扩增Treg细胞,在小鼠模型中实现白癜风长期缓解,局部雷帕霉素治疗的临床试验正在开展;CD25靶向IL-2变体可选择性扩增Treg细胞,在健康受试者中证实有效,目前已进入白癜风患者的II期临床试验。此外,中和IL-15、靶向CD122等干预记忆T细胞的策略,也处于临床开发阶段。


随着JAK抑制剂等批准药物的面市,白癜风的临床治疗进入了靶向免疫调控的新时代。过去四十年的研究已系统揭示了白癜风的免疫发病机制,明确了CD8+ T细胞、IFNγ-CXCR3轴及TRM在疾病发生、持续及复发中的核心作用,也证实了先天免疫激活、Treg功能异常的始动及调控作用。
未来研究的核心方向为开发高选择性的免疫干预策略,在阻断黑色素细胞破坏的同时避免广泛的免疫抑制;深入解析皮肤微环境中细胞间的相互作用,明确TRM的调控机制,有望为疾病复发提供新的干预靶点。此外,白癜风与黑色素瘤免疫治疗的交叉研究,不仅为白癜风提供了新的研究思路,也将反哺对CD8+ T细胞自身抗原耐受及记忆的基础认知,为自身免疫病与肿瘤免疫治疗的协同发展提供助力。
参考文献:
医脉通是专业的在线医生平台,“感知世界医学脉搏,助力中国临床决策”是平台的使命。医脉通旗下拥有「临床指南」「用药参考」「医学文献王」「医知源」「e研通」「e脉播」等系列产品,全面满足医学工作者临床决策、获取新知及提升科研效率等方面的需求。

(本网站所有内容,凡注明来源为“医脉通”,版权均归医脉通所有,未经授权,任何媒体、网站或个人不得转载,否则将追究法律责任,授权转载时须注明“来源:医脉通”。本网注明来源为其他媒体的内容为转载,转载仅作观点分享,版权归原作者所有,如有侵犯版权,请及时联系我们。)








