编者按

大疱性表皮松解症(EB)是一组以皮肤黏膜脆性增加、轻微外伤后易出现水疱或糜烂为特征的遗传性皮肤病,依据水疱发生部位可分为多个亚型。其中,痒疹型


(一)主诉
全身反复发作剧烈瘙痒性
(二)现病史
患者女性,40余岁。因“全身反复发作剧烈瘙痒性皮疹30年”就诊于我院自身免疫性大疱病专科门诊。皮疹主要分布于双下肢、前臂和手部。患者自诉几乎终生存在轻微外伤后皮肤即易剥脱的现象,但无黏膜受累史。曾多次在外院接受治疗,包括外用
(三)既往史与外院检查
外院活检结果提示:血管周围及局灶性苔藓样淋巴细胞组织细胞浸润,伴粟丘疹及表皮下分离,曾怀疑获得性大疱性表皮松解症或人为分离。
(四)体格检查
体检可见其双小腿前侧、足背及前臂伸侧遍布数百个棕色至紫红色的鳞屑性、苔藓样丘疹(图A),部分呈线状排列,上、背部有少量散在
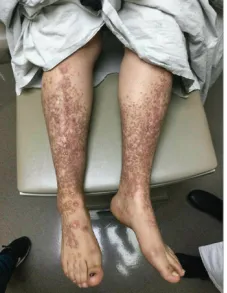
图1 下肢皮损临床表现:患者双侧下肢及足背散在数百个2-5毫米大小的红褐色平顶丘疹。同时可见大拇趾甲营养不良
(五)辅助检查
为明确诊断,我们进行了以下检查:
1.组织病理学检查(HE染色):显示局灶性苔藓样皮炎伴真皮-表皮交界处表皮下分离及真皮纤维化,未见空泡变性或嗜酸性粒细胞浸润。同时存在角化过度、棘层增厚和表皮萎缩区域(图B)。
2.直接免疫荧光检查:结果显示基底膜带无免疫反应物线性沉积。
3.血清学检查:VII型胶原抗体
4.基因检测:COL7A1基因存在c.7247G>T序列变异杂合子。
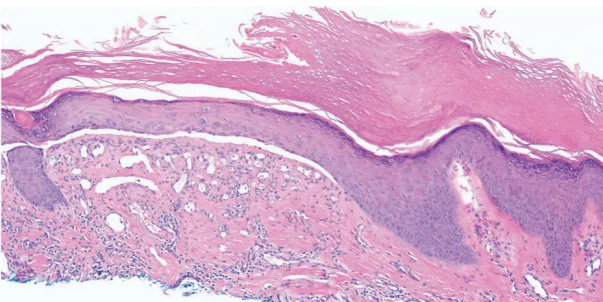
图2 皮损组织病理学图像(HE染色,×10):篮网状角化过度,其下方表皮呈棘层增厚与萎缩相间,稀疏的淋巴细胞组织细胞围管性皮炎,伴表皮下分离及真皮纤维化。


(一)最终诊断
营养不良型大疱性表皮松解症-痒疹型(DEB-P)
(二)诊断依据
1.临床表现:长达30年的慢性病程、剧烈且难治的瘙痒、四肢(尤其小腿)特征性的苔藓样平顶丘疹、皮肤脆性增加及甲营养不良。
2.组织病理:提示表皮下分离。
3.实验室检查:直接免疫荧光和VII型胶原抗体检测排除了获得性大疱性表皮松解症。
4.基因检测:发现COL7A1基因杂合突变,COL7A1基因编码VII型胶原(锚原纤维主要成分),其突变是DEB-P的致病根源,结合痒疹样临床表现确诊为DEB-P。
(三)鉴别诊断
本病例曾与以下疾病进行鉴别:
1.苔藓样淀粉样变:组织病理中未见显著的淀粉样物质沉积,不支持此诊断。
2.扁平苔藓:本例病理缺乏致密的苔藓样浸润、空泡变性、锯齿状上皮脚及胶样小体,且病程长达30年,均不符合扁平苔藓的典型表现。
3.结节性痒疹:重复活检显示明确的表皮下分离,不支持单纯的结节性痒疹。
4.获得性大疱性表皮松解症:虽然组织学上的表皮下分离和稀疏炎性浸润与之相符,但直接免疫荧光阴性、血清学抗体阴性以及临床上无水疱,有力地排除了该诊断。


确诊后,患者初始服用加巴喷丁治疗,瘙痒得到部分缓解。随后,医生为其加用了


营养不良型大疱性表皮松解症-痒疹型是一种于1994年被描述的罕见DEB亚型,由编码VII型胶原(锚原纤维的主要成分)的COL7A1基因发生多种变异所致。本病通常为常
(一)临床特征
DEB-P患者常不出现典型水疱,而以剧烈瘙痒和下肢为主的苔藓样丘疹、结节为突出表现,皮损可融合成斑块,常呈线状排列,极易与苔藓样淀粉样变或扁平苔藓混淆。皮肤脆性增加、瘢痕、粟丘疹和甲营养不良是常见伴随症状。本病可在出生时即发病,也可延迟数十年,这增加了误诊的风险。
(二)病理与机制
其病理表现多样,通常包括角化过度、棘层增厚、表皮下分离及以单核细胞为主的血管周围浸润。本病例中观察到的稀疏炎性浸润和表皮下分离是支持DEB诊断的关键线索。剧烈瘙痒的机制尚不完全清楚,可能与Th1、Th2、Th17细胞因子通路失调、皮肤屏障功能受损以及表皮感觉神经异常活化有关。本例患者使用度普利尤单抗(一种抗IL-4Rα全人源单克隆抗体,可同时抑制IL-4/IL-13信号通路)疗效显著,提示Th2通路在发病中起重要作用。
综上,对于表现为慢性、顽固性、非水疱性瘙痒丘疹的患者,即使成年后才出现典型症状,也应将DEB-P纳入鉴别诊断。全面的临床评估结合组织病理学、免疫荧光及基因检测,是避免误诊、实现精准诊断的关键。
参考文献:
医脉通是专业的在线医生平台,“感知世界医学

(本网站所有内容,凡注明来源为“医脉通”,版权均归医脉通所有,未经授权,任何媒体、网站或个人不得转载,否则将追究法律责任,授权转载时须注明“来源:医脉通”。本网注明来源为其他媒体的内容为转载,转载仅作观点分享,版权归原作者所有,如有侵犯版权,请及时联系我们。)








