

1细胞焦亡
目前认为,细胞焦亡的发生主要有2种分子通路介导,一种是炎症小体激活Caspase-1经典途径,另一种是脂多糖直接激活Caspase-4/5/11的非经典途径。而炎症性半胱天冬酶(Caspase-1、Caspase-4、Caspase-5、Caspase-11)和消皮素(GSDM)蛋白家族成员,是细胞焦亡执行其效应功能所必需的核心分子。
炎症小体是细胞应答微生物感染产生的微生物相关分子模式和/或受损或死亡细胞释放的内源性危险信号损伤相关分子模式,在细胞质中组装的一种大型多蛋白复合物。经典炎症小体主要由感受器蛋白、衔接蛋白和效应蛋白构成:感受器蛋白主要由NLR蛋白家族和含有HIN结构域的蛋白家族如热蛋白-HIN结构域蛋白组成;衔接蛋白是凋亡相关斑点样蛋白,可介导感受器蛋白和效应蛋白间的相互作用;效应蛋白通常是指Pro-Caspase 1。此外,非经典炎症小体在多种疾病进程中可能发挥着重要作用,其机制涉及人源Caspase-4、Caspase-5及鼠源Caspase-11的参与,这些炎症性半胱天冬酶兼具感受器蛋白与效应蛋白的双重作用。炎症小体作为炎症性半胱天冬酶活化平台,通过激活该类蛋白酶进而引发焦亡,最终导致溶解性细胞死亡并伴随炎症介质的释放。
细胞焦亡过程中,活化的炎症性半胱天冬酶可特异性裂解GSDM蛋白,后者作为成孔效应因子在细胞膜上形成孔道,是执行细胞焦亡的关键因子。人类GSDM家族包含6个成员,即GSDMA、GSDMB、GSDMC、GSDMD、GSDME和GSDMF,而在小鼠中缺失GSDMB,其GSDMA与GSDMC则存在多种亚型。除GSDMF外,大多数GSDM蛋白均由以下3个结构域构成:GSDM-N(N末端结构域)、连接区及GSDM-C(C末端结构域)。在应答激活信号时,GSDM蛋白可被不同的炎症性半胱天冬酶在中央连接区进行切割。通常GSDM-C对GSDM-N发挥抑制作用,故其过表达可抑制细胞死亡;而一旦GSDM-N摆脱抑制,即可与膜脂结合并大量成孔,导致细胞毒性及死亡。此外,活化的炎症性半胱天冬酶还可水解白细胞介素(IL)-1β、IL-18等炎症因子前体,使其转变为成熟且有生物活性的形式。经GSDM蛋白切割后形成膜孔,炎症因子得以通过膜孔释放至胞外,进而参与免疫应答并触发炎症级联反应。

2细胞焦亡在肝纤维化中的作用
越来越多的研究表明,细胞焦亡在多种肝疾病发病机制中具有重要作用。在各类炎症小体中,以NLRP3炎症小体的研究最为广泛,调控NLRP3活性被认为是治疗肝疾病的潜在策略。研究表明,在NASH小鼠模型中,NLRP3表达上调伴随肝脏炎症与纤维化程度加重,而使用NLRP3抑制剂MCC950不仅可有效抑制NLRP3活性,还能显著缓解肝脏炎症和纤维化进程。除NLRP3炎症小体外,研究显示,干扰素激活基因2蛋白(AIM2)、NLRP6等其他炎症小体亦参与肝纤维化进程。GSDM蛋白作为细胞焦亡发生过程中的关键蛋白之一,Zhu等在代谢功能障碍相关脂肪性肝炎小鼠模型中观察到,GSDME基因敲除可减轻肝脏病理损伤。以下对细胞焦亡参与肝纤维化的主要作用机制及研究进展进行总结。
HSC状态与功能的改变是肝纤维化过程的中心环节,已有许多研究证实细胞焦亡与之密切相关。
NLRP3炎症小体在HSC活化过程中具有重要作用。研究表明,在特异性诱导HSC内NLRP3炎症小体过度活化的小鼠模型中,其肝纤维化程度加重,并可促进HSC表型向肌成纤维细胞转化。Li等发现醛固酮可通过上调NLRP3炎症小体的表达促进原代小鼠HSC活化,而在NLRP3基因敲除的小鼠中,醛固酮诱导的肝纤维化程度显著减轻,说明NLRP3炎症小体表达能够推动HSC活化进而促进肝纤维化进展。在甘氨鹅脱氧
细胞焦亡所释放的炎症因子对HSC活化具有重要的调控作用。NLRP3炎症小体活化后产生的IL-1β可直接或间接激活HSC,并上调纤维化标志物表达水平;而IL-18亦被证实通过促进HSC活化推动肝纤维化进展。肝硬化患者血清中的IL-18及其结合蛋白IL-18 BP水平显著升高,在四氯化碳诱导的肝纤维化小鼠模型中,活化的HSC高表达IL-18及其受体IL-18R1。动物实验进一步表明,IL-18可促进小鼠HSC活化,诱导α-SMA及相关纤维化基因表达上调,而使用IL-18拮抗剂则能显著抑制上述效应,提示抑制IL-18可能成为治疗肝纤维化的一种潜在策略。Xie等研究表明,
上述研究表明,在肝纤维化进程中,HSC的焦亡主要通过损伤因子激活的NLRP3炎症小体介导的两种通路调控:经典途径为NLRP3炎症小体激活Caspase-1,裂解GSDMD形成膜孔并释放IL-1β/IL-18;非经典途径则由Caspase-4/5直接裂解GSDMD诱导焦亡。释放的IL-1β与IL-18通过旁分泌作用激活周围HSC,上调α-SMA等纤维化标志物表达,从而促进胶原沉积与纤维化进展。
肝实质细胞损伤是肝疾病进展的关键初始事件,其死亡后释放的损伤相关分子模式可向周围细胞传递危险信号,在肝脏炎症与纤维化发展中具有重要作用。肝实质细胞发生焦亡对肝纤维化进程具有重要影响。
已有研究通过构建全身性和髓系细胞特异性NLRP3敲入小鼠模型,探究肝实质细胞焦亡机制,结果显示,全身性NLRP3敲入小鼠出现更显著的肝实质细胞焦亡及肝纤维化程度。肝实质细胞中NLRP3过度激活可诱导其发生焦亡,并将NLRP3炎症小体释放至胞外,进而被HSC内吞并促进活化,最终推动肝纤维化进展。研究还发现,在四氯化碳诱导的肝纤维化小鼠模型肝组织中生长阻滞特异性转录因子5(GAS5)表达显著下调,而过表达GAS5可下调α-SMA表达并抑制Ⅰ型胶原蛋白mRNA表达。为进一步阐明GAS5的作用机制,分离培养肝实质细胞与HSC,结果证实,GAS5可通过抑制肝实质细胞焦亡,从而抑制HSC活化,延缓肝纤维化进程。Xiao等研究发现,干扰素基因刺激因子(STING)可通过招募WD重复结构域5与DOT1样组蛋白H3K79甲基转移酶促进组蛋白甲基化,增强干扰素调节因子3与NLRP3启动子区域结合,从而上调NLRP3表达。
由此可见,NLRP3炎症小体介导的焦亡是肝实质细胞损伤及继发HSC活化、进而推动肝纤维化的重要机制。现有研究提示,靶向抑制STING-NLRP3-GSDMD焦亡轴或上调GAS5表达,可能成为治疗肝纤维化的潜在靶点。
肝巨噬细胞属于肝脏非实质细胞群,主要包括自我更新的组织驻留型肝巨噬细胞以及由腹腔和骨髓募集的巨噬细胞。该类细胞在维持系统稳态、应答感染、启动肝损伤炎症反应、推动肝纤维化进展、促进伤口愈合及肝纤维化逆转等过程中发挥着重要作用。
在肝纤维化过程中,细胞焦亡与肝巨噬细胞功能密切相关。Shu等研究表明,METTL3/MALAT1/PTBP1/USP8/TAK1信号通路在肝纤维化过程中被激活,可促进肝巨噬细胞发生焦亡并极化为M1表型,从而加剧肝纤维化进展。Wan等发现熊果酸对四氯化碳诱导的小鼠肝纤维化具有保护作用,其机制是通过抑制烟酰胺腺嘌呤二核苷酸磷酸氧化酶(NOX)2介导的NLRP3炎症小体活化,减少炎症介质的释放,抑制肝巨噬细胞焦亡,进而减缓肝纤维化进程。Liu等报道S100钙结合蛋白A8(S100A8)通过激活TLR4/NF-κB信号通路并诱导ROS生成,促进NLRP3依赖性肝巨噬细胞焦亡,从而推动肝纤维化进程。此外,该研究还提示,与细胞焦亡相关的血清GSDMD水平对肝纤维化诊断与分期具有潜在价值,提示该指标可能成为评估肝纤维化的生物标志物。另有研究指出,肝巨噬细胞与中性粒细胞胞外陷阱(NET)-AIM2炎症小体驱动肝纤维化进展这一过程相关,研究认为NET可被肝巨噬细胞吞噬,其双链DNA成分激活AIM2炎症小体,诱导肝巨噬细胞焦亡并释放IL-1β、IL-18等炎症因子,进而促进HSC活化,诱导肝纤维化发生。
综上所述,在肝纤维化进程中,肝巨噬细胞焦亡的核心调控机制涉及多条信号通路:S100A8/TLR4/NF-κB-ROS-NLRP3通路、METTL3/MALAT1/PTBP1/USP8/TAK1通路、NET-AIM2通路以及NOX2/NLRP3通路均可促进肝巨噬细胞发生焦亡,并通过释放IL-1β/IL-18持续活化HSC,进而驱动胶原沉积与纤维化进程的恶性循环。针对以上通路进行干预(例如熊果酸通过抑制NOX2/NLRP3通路发挥作用),可能成为调控肝巨噬细胞焦亡、缓解肝纤维化的潜在治疗策略。
众多研究表明,肝脏DC参与肝纤维化的发生发展。在曼氏血吸虫感染小鼠模型中,虫卵及其可溶性虫卵抗原可刺激肝脏DC,使其通过激活NLRP6炎症小体引发细胞焦亡,焦亡所产生的炎症因子进一步活化HSC并推动肝纤维化进程;且NLRP6基因敲除小鼠感染曼氏血吸虫后,其肝脏肉芽肿面积和胶原沉积显著减少,纤维化相关标志物水平也明显下降。LSEC的焦亡亦能加剧肝纤维化程度。Yu等研究发现,在

3小结
肝脏中的肝实质细胞、HSC、肝巨噬细胞以及其他类型肝细胞发生的细胞焦亡均被证实可促进肝纤维化的发生发展(图1)。作为肝纤维化进程中的核心效应细胞,HSC不仅自身焦亡可直接加剧纤维化,肝实质细胞与肝巨噬细胞等其他肝细胞焦亡后所产生的标志物(如NLRP3炎症小体、IL-1β、IL-18)亦可作为关键信号分子驱动HSC活化,进而推动纤维化进程。目前研究多聚焦于NLRP3介导的经典焦亡途径,其抑制剂MCC950已显示出抗纤维化潜力;此外,针对AIM2、NLRP6其他炎症小体在肝纤维化中作用的研究也表明,靶向抑制这些炎症小体可能成为干预肝纤维化的新策略。然而,将调控细胞焦亡的策略转化为肝纤维化临床治疗方案,仍面临以下亟需解决的关键科学问题:一方面,细胞焦亡在固有免疫防御中具有重要的防御作用,广泛抑制炎症小体可能削弱机体免疫监视功能,因此,需精准靶向慢性肝病中特定肝细胞的异常炎症小体活化,以期在抑制致病性焦亡的同时,最大程度维持全身免疫稳态;另一方面,细胞焦亡所释放的IL-1β、IL-18等炎症因子是直接激活HSC、驱动纤维化的核心效应分子,开发特异性拮抗HSC表面相应受体(如IL-1R、IL-18R)或干扰其信号转导的药物,可抑制炎症因子过度活化HSC,完善靶向治疗的关键环节。综上,深入探索肝纤维化发生发展中细胞焦亡的特异性机制,并发展安全、精准的干预策略,是未来焦亡靶向治疗取得突破的关键所在,但其临床转化仍然任重道远。
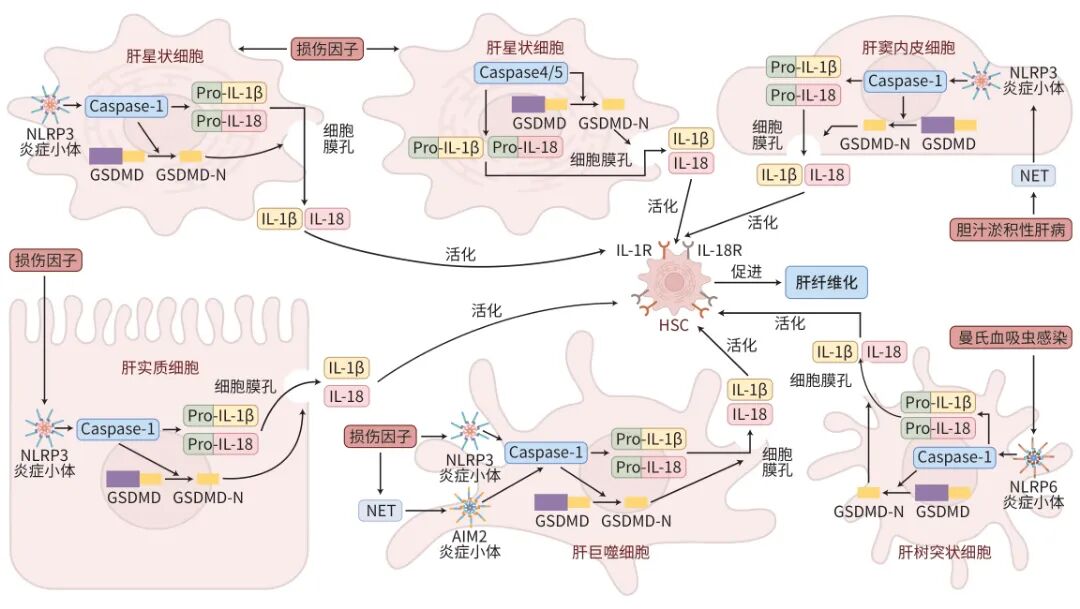
注: NLRP3,NOD样受体蛋白3;GSDMD,消皮素D;GSDMD-N,消皮素D的N端结构;NET,中性粒细胞胞外陷阱;AIM2,干扰素激活基因2蛋白;IL-1β,白细胞介素1β;IL-18,白细胞介素18;Pro-IL-1β,白细胞介素1β前体;Pro-IL-18,白细胞介素18前体。
图1 不同肝细胞焦亡促进肝纤维化的机制图

https://www.lcgdbzz.org/cn/article/doi/10.12449/JCH260125

金泳帆, 赵春梅, 邰文琳. 细胞焦亡在肝纤维化中的作用及研究进展[J]. 临床肝胆病杂志, 2026, 42(1): 197-202
来源:临床肝胆病杂志
本平台旨在为医疗卫生专业人士传递更多医学信息。本平台发布的内容,不能以任何方式取代专业的医疗指导,也不应被视为诊疗建议。如该等信息被用于了解医学信息以外的目的,本平台不承担相关责任。本平台对发布的内容,并不代表同意其描述和观点。若涉及版权问题,烦请权利人与我们联系,我们将尽快处理。
(本网站所有内容,凡注明来源为“医脉通”,版权均归医脉通所有,未经授权,任何媒体、网站或个人不得转载,否则将追究法律责任,授权转载时须注明“来源:医脉通”。本网注明来源为其他媒体的内容为转载,转载仅作观点分享,版权归原作者所有,如有侵犯版权,请及时联系我们。)








