(1)IgG4的来源?有何特点?
(2)IgG4相关性疾病(IgG4-relateddisease,IgG4-RD)病名的由来?怎样发病?
(3)IgG4-RD的流行病学和临床表现?
(4)实验室检查有何特点?有何特征性的病理变化?
(5)如何确诊?与Castleman病如何鉴别?
(6)怎样治疔?
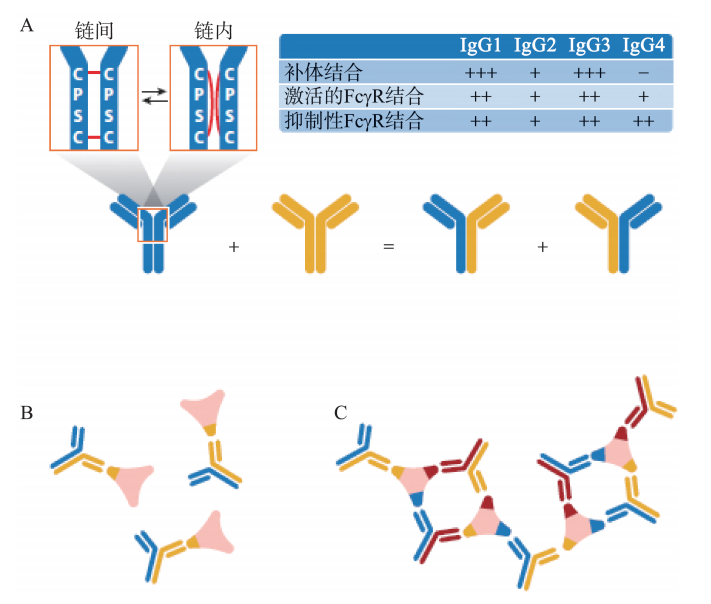
正常B细胞的成熟经历了前B细胞、不成熟B细胞、成熟B细胞、活化B细胞和浆细胞5个阶段。其中前B细胞和不成熟B细胞的分化是抗原非依赖的,其分化过程在骨髓中进行。抗原依赖阶段是指成熟B细胞在抗原刺激后活化,并继续分化为合成和分泌抗体免疫球蛋白(Ig)的浆细胞。Ig有5种,即IgG、IgA、IgD、IgM和IgE。IgG有1~4四种亚型,IgG4亚型的特点是其IgG4的Fab臂与另一个IgG4的Fab臂交换,形成双特异的抗体和很大的免疫复合物,引起组织病理变化。IgG4是一种非炎症性抗体。Fab臂交换、无法结合补体、与激活的FcyRI结合不良以及与抑制性FcγR(FcγRIIB)结合的能力是IgG4的独特特征,这些特征使其成为非炎症性免疫球蛋白。IgG4的重链可以在链间和链内二硫键结构之间切换。链内二硫键连接的IgG4由非共价结合的半IgG4分子(可在体外、还原条件下或体内进行交换)组成,可能受到FcRn再循环的协助。这一过程称为Fab臂交换,可产生双特异性IgG4抗体,形成免疫复合物。在IgG4相关性疾病中,体内形成的免疫复合物的性质尚不清楚。有两种可能性:①抗原特异性IgG4与无关特异性抗体的Fab臂交换可能导致形成功能性单价抗体。这种抗体会产生小型非沉淀性免疫复合物。②与同一抗原上不同表位反应的IgG4抗体之间的Fab臂交换将产生功能性二价抗体,并可能导致形成很大的免疫复合物。由于缺乏补体结合,此类免疫复合物可能无法有效清除,并且可能是某些IgG4相关性疾病病例中出现IgG4沉积的原因(图48-1)。
图48-1 IgG4亚型的特点
A.IgG4-Fab臂交换产生双特异性抗体;B.具有功能性单价的双特异性抗体:非沉浸免疫复合物;C.无功能性单价的双特异性抗体:潜在的大免疫复合物
IgG4-RD是一种免疫介导的慢性纤维化炎性病理过程,有向各器官发展形成肿瘤样病变的趋向,可以按同步或非同步的方式发生。IgG4-RD在各器官的病理表现已在19世纪通过病理组织检查确定。过去有许多疾病现已知属于IgG4-RD(表48-1),它们的病理基础是相同的,如鼻硬结病、Riedel甲状腺炎、MorbusOrmond病或Küttner肿瘤等,既往都被认为是罕见的、孤立的疾病。1995年,对IgG4-RD发现的第一步是对一种自身免疫介导、激素敏感型的胰腺炎的描述,今天被称为IgG4-RD或1型自身免疫性胰腺炎(AIP)。随后,2001年,Hamano等报道血清IgG4水平升高的AIP患者和特征性组织病理学模式合并腹膜后纤维化(RPF),从而真正认识了IgG4-RD多器官表现。2012年,对IgG4-RD的统一命名发表,放弃了IgG4-RD及其器官表现的所有其他同义名称。在同一年,日本工作组介绍IgG4-RD诊断标准,并对IgG4-RD病理学诊断达成国际共识。
表48-1以下一些过去已知的疾病,现已归入IgG4-RD
IgG4-RD是一个非常年轻的病种。虽然人们普遍认为是一种罕见的疾病,但随着越来越多的认识和研究进展,国内已有不少报道,相信随着临床医生诊断水平的提高,诊断为IgG4-RD的患者数量将会增加。
IgG4-RD的发病机制尚未完全明了,可能与下述因素有关。
正常人IgG4的血清含量为60mg/dl,占所有IgG的3%,补体结合能力较低,可透过胎盘,主要攻击靶点为寄生虫和多糖体。由于在铰链区氨基酸变异,IgG4分子可以分裂,并随机结合其他的半分子(Fab臂交换),与两个不同的抗原结合位点产生不对称的双特异性抗体。这种特殊的功能容易形成很大的免疫复合物,由于IgG4结合补体的亲和力低,因此,此免疫复合物不易被清除,启动免疫反应的能力有限且可在某些IgG4-RD的病理组织中沉积,IgG4是否直接致病或只是伴发于疾病,目前还不清楚。
由于IgG4-RD的发现,一个2型辅助性T细胞(Th2)驱动的免疫机制在其发病机制中的研究已经在进行。Th2型细胞因子,如IL-4、IL-5和IL-13,以及T细胞调节的相关因子如IL-10和转化生长因子-β(TGF-β)存在于IgG4-RD中。IL-13、TGF-β可能通过激活成纤维细胞导致纤维化,而IL-4和IL-10被认为引起B淋巴细胞的特异性IgG4类开关重组导致此亚型浆细胞增加。然而,这个模型的IgG4-RD远远不能解释该病的所有特征。
最新也有研究质疑IgG4-RD模型作为一个主要的T-细胞驱动的疾病。在免疫组化研究中,肥大细胞而不是T细胞似乎产生了IL-4、L-10和TGF-β。
总结起来,天生免疫在lgG4-RD发病机制中的作用在过去被低估了,需要进一步研究闸明。一样的问题,IgG4抗体代表致病因素或只是疾病的伴随因索也需进一步研究。其他重要的研究目标是可能的抗原触发了IgG4一RD的发生和B细胞以及T细胞的相互作用,包括最终导致纤维化的机制。
到目前为止,关于IgG4-RD及其器官表现的流行病学知道得很少。目前还不清楚是否亚洲人比他洲的人易感IgG4-RD,目前,现有的流行病学数据主要来自日本的数据。Uchida等人估计IgG4-RD的年发病率在(0.28~1.08)/100000,2009年,共有8000名患者在日本发病,发病率为62例/100万人口。
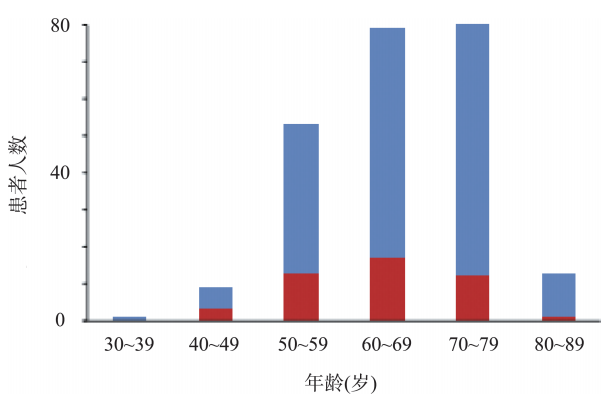
IgG4-RD通常更容易影响中老年,50~70岁发病概率大(图48-2),罕见有儿科病例的描述。大多数研究报告显示整体IgG4-RD好发于男性,尤其是IgG4相关性胰腺炎(男女比为7:3)。然而,IgG4相关的涎腺炎和泪腺炎可能更好发于女性。
图48-2 235例患者的发病年龄
IgG4-RD患者的年龄分布。200例患者(91%)的年龄在50~80岁。蓝色和红色条分别代表男性和女性患者
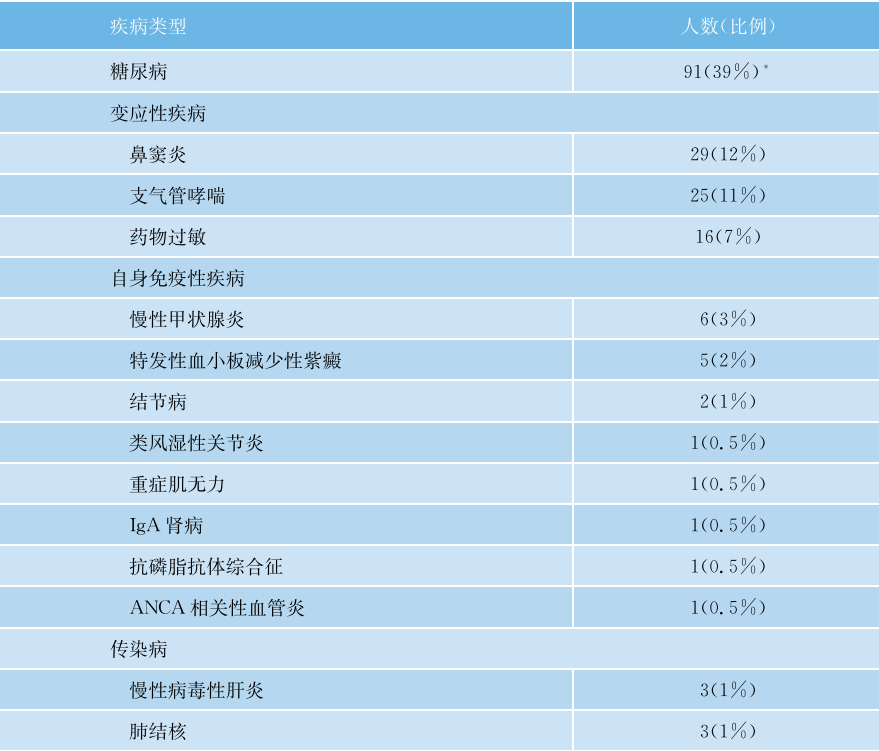
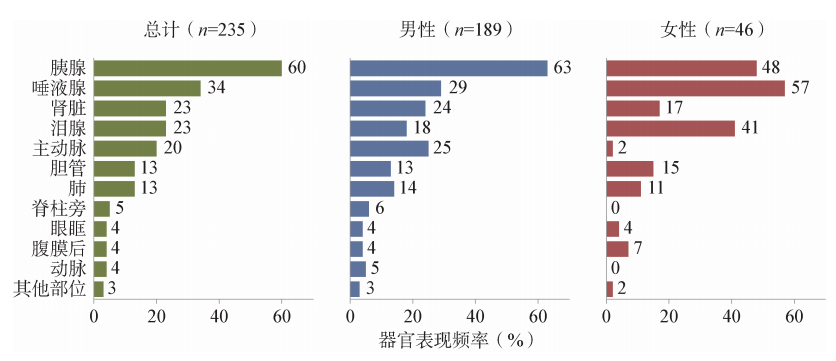
在日本的一个研究中,据报道70%的患者存在IgG4-RD的症状,而30%的患者属于意外发现。最频繁的症状与肿胀相关,如阻塞性黄疽、眼球突出(41%),其次是腹部症状(18%)、乏力(4%)。肾(4%)和肺部症状(3%)只出现在少数患者中。另一个常见的诊断前症状是体重减轻。在IgG4相关性胰腺炎患者,腹痛(65%)、黄疽(62%)和体重减轻(42%)是最常见的症状。表48-2为一组235例患者既往所患疾病,图48-3为这些患者的病灶累及的脏器。
图48-2 235例患者的既往疾病
*63例患者表现为1型AIP
图48-3 235例IgG4-RD患者诊断时不同器官的累及情况。其他部位包括前列腺、周围神经、垂体、皮肤和心包
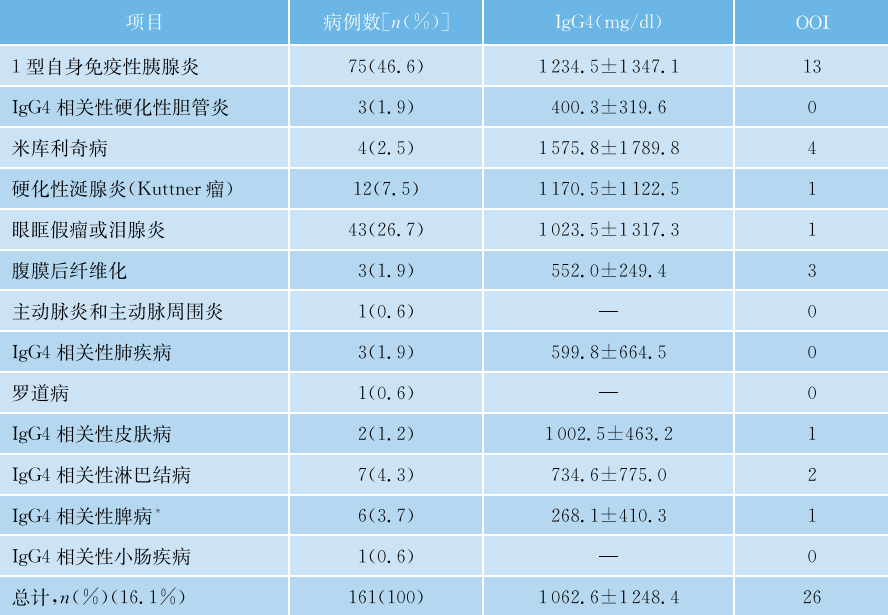
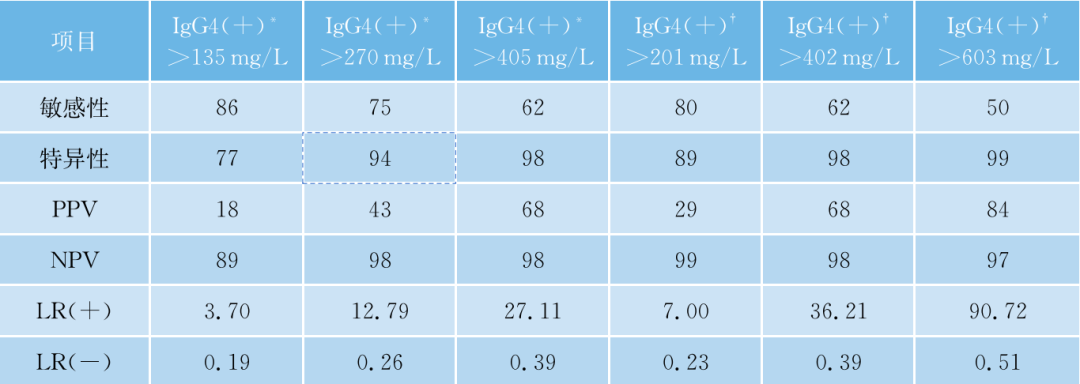
中国台湾学者的相关研究对2901例病例的血清IgG4进行了检测,其中包含161例IgG4-RD和2740例非IgG4相关性疾病。135mg/dl(1.35g/L)是比较好的诊断临界值(表48-3、表48-4)。
表48-3 161例根据累及器官和组织分层的中国IgG4-RD患者的临床表现和血清IgG4浓度
OOI=胰腺外器官受累;*硬化性血管瘤样结节转化
表48- 4 不同血清IgG4临界值分层在IgG4-RD和非IgG4-RD的患者中的表现
LR(-),阴性似然比,LR(+),阳性似然比,NPV,阴性预测值,PPV,阳性预测值。
*大多数之前的研究确定的IgG4临界值(135mg/dl);†根据生产商的药品说明书定义的IgG4临界值(3~201mg/dl)
活检和组织学证据是IgG4-RD诊断的主要标准,有条件应尽量获得。免疫组化染色和IgG4单克隆抗体可以帮助诊断。三大主要病理学表现,必须至少存在两个,包括:①密集的淋巴浆细胞浸润;②席纹状纤维化;③闭塞性静脉炎。
关于免疫组化,尽管不同器官的特异性阈值有差异,>50IgG4(+)浆细胞每高倍视野通常被视为IgG4-RD高度特异性的。然而,在高度炎症或纤维化的患者,对应的浆细胞数量明显增加或极其缺乏,计数IgG4(+)浆细胞绝对数是不可靠的。因此,建立了IgG4(+)浆细胞和IgG+浆细胞的比值,以>40%作为诊断临界值,其敏感性为94.4%,特异性为85.7%。就像血清IgG4,IgG4(+)浆细胞不仅存在于IgG4-RD,还可能存在于其他炎症或肿瘤性疾病。因此,对IgG4-RD的诊断不能单独基于IgG4(+)浆细胞的存在,还必须同时伴随典型的IgG4-RD组织学外观和密切的临床相关性。
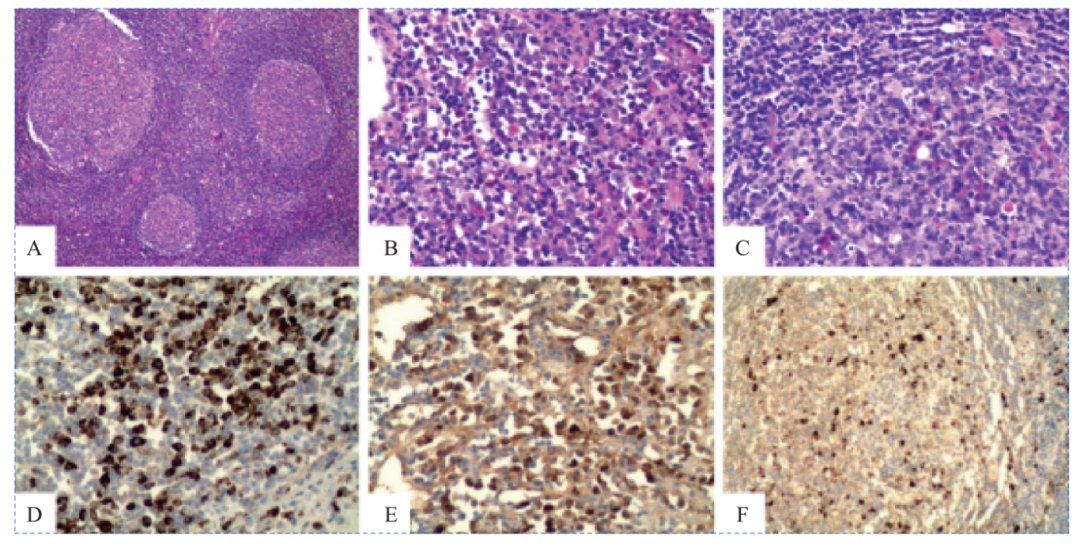
图48-4 IgG4相关性淋巴结病的病理表现
右侧颌下淋巴结肿大显示滤泡增生型IgG4相关性淋巴结病。A.可见大量反应性淋巴滤泡(原始放大倍数×40)。B.滤泡间区的嗜酸性粒细胞和成熟浆细胞增多(原始放大倍数×200)。C.生发中心内可见成熟浆细胞明显浸润(原始放大倍数×200)。IgG4(D)和IgG(E)的免疫染色显示出大量IgG4阳性细胞(130/高倍视野和超过70%的IgG4/IgG比率,原始放大倍数×200)。F.生发中心内IgG4阳性细胞增多(原始放大倍数×200)
表48-5为IgG4-RD诊断标准。结合本患者,有免疫相关的临床表现、消瘦及严重低蛋白血症,影像学见多发肿大淋巴结,IgG4 15.50g/L,显著超过诊断临界值,淋巴结病理有类似Castleman病的表现,加做IgG4标记,阳性率超过40%,结合三方面的依据,可以确诊为IgG4相关淋巴结病。
表48-5 IgG4-RD诊断标准
注:数据来自Okazaki、Umehara和Umehara等。
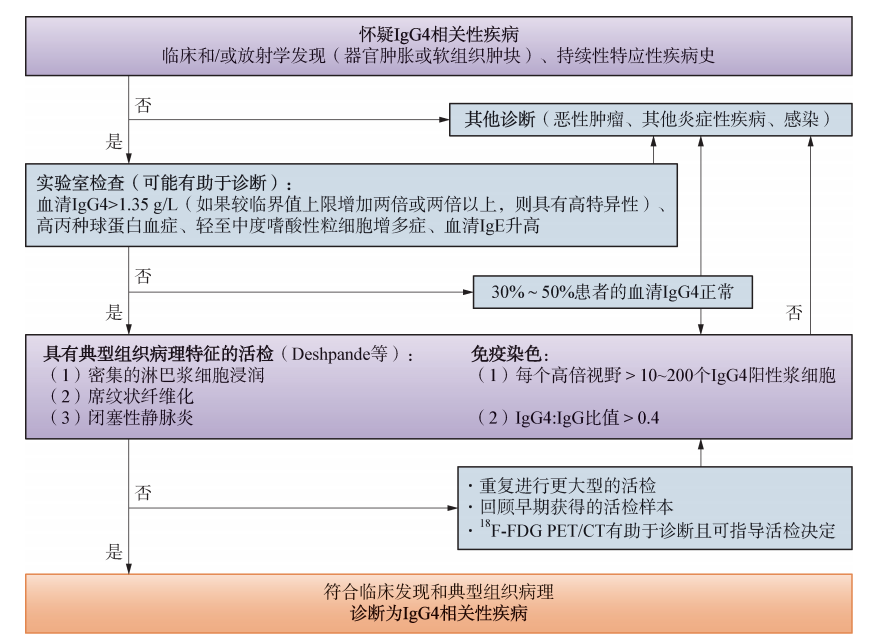
图48-5是最近国际会议上议定的诊断IgG4-RD的诊断流程。疑有IgG4-RD时,血清IgG4测定(>1.35g/L)、高球蛋白血症,病理切片中,在高倍镜下,IgG4阳性浆细胞10~200个是重要诊断依据。本病例确诊前曾有反复和争论,最后在病理切片中发现IgG4阳性浆细胞超过40%,才排除Castleman病,而确定为IgG4相关淋巴结病。
图48-5诊断流程图
 我要投稿
我要投稿

















{kind=link}