
近期发表于Alzheimer’s & Dementia的一篇综述围绕“2型糖尿病如何改变阿尔茨海默病风险”进行了系统梳理,并结合人类死后脑组织分子数据重新分析,提出了一个更接近当前证据的新解释:2型糖尿病相关的外周脂质异常和炎症可能共同削弱血脑屏障完整性,促进外周与中枢之间异常免疫信号交流,进而诱导与阿尔茨海默病相关的小胶质细胞亚型改变;这些小胶质细胞脂质代谢异常,可能比“中枢

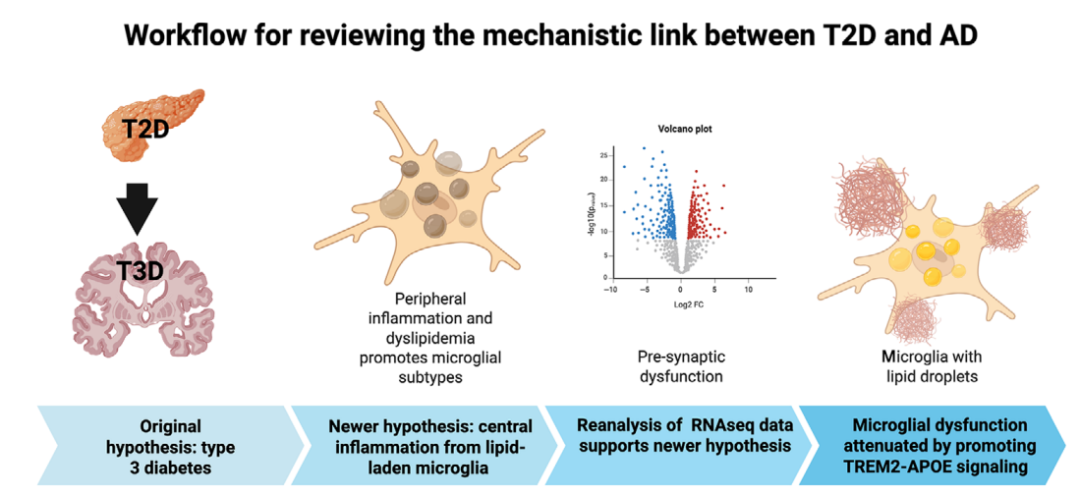
2型糖尿病与AD并非简单相关
阿尔茨海默病是最常见的痴呆类型,其核心病理改变包括细胞外β淀粉样蛋白(Aβ)斑块、神经元内神经原纤维缠结,以及反应性胶质细胞、神经炎症和血管改变。随着疾病进展,神经原纤维缠结在脑内扩散,其范围和程度与疾病严重程度密切相关,最终导致神经元死亡和临床症状出现。
从公共卫生角度看,2型糖尿病已被纳入痴呆的可干预危险因素之一。Lancet Commission相关报告提出,14项可干预生活方式和健康因素合计可解释约45.3%的痴呆风险,其中糖尿病在总体人群中的归因风险比例约为2%。这一数字低于高LDL胆固醇和听力损失等因素,但由于2型糖尿病患病人数庞大,其对认知健康的长期影响仍不容忽视。
过去很长一段时间,2型糖尿病与阿尔茨海默病之间的联系常被解释为“中枢胰岛素抵抗”。这一假说认为,阿尔茨海默病可能类似发生在脑内的糖尿病,因此也曾被称为“3型糖尿病”。支持这一假说的理由包括:胰岛素信号通路与糖原合成酶激酶3β(GSK3β)等分子相关,而GSK3β也参与tau蛋白磷酸化;糖尿病中的胰岛素抵抗、氧化应激和炎症反应,似乎都能与阿尔茨海默病病理发生联系。
但新的证据正在使这一解释变得不再充分。脑并不是典型的胰岛素敏感器官。
因此,2型糖尿病增加阿尔茨海默病风险这一事实仍然成立,但其核心机制可能并不是“脑内胰岛素失灵”这么简单。
糖尿病影响认知,血管通路仍是重要背景
糖尿病与痴呆之间的流行病学联系并不弱,但其对不同类型痴呆的影响并不完全一致。基于UK Biobank数据的研究曾显示,糖尿病与全因痴呆、阿尔茨海默病和
这并不令人意外。2型糖尿病可通过
不过,单纯用“血管损伤”解释糖尿病与阿尔茨海默病之间的关系仍然不够。对UK Biobank数据的再分析显示,中年期糖尿病与全因痴呆风险增加相关,调整后HR为1.14(95%CI:1.02–1.27,P=0.018);但在老年期糖尿病比较中,这一关联不再显著,调整后HR为1.11(95%CI:0.96–1.30,P=0.15)。这一结果提示,糖尿病对脑健康的影响可能具有时间窗口,中年期长期代谢异常或许更容易参与后续认知风险累积。
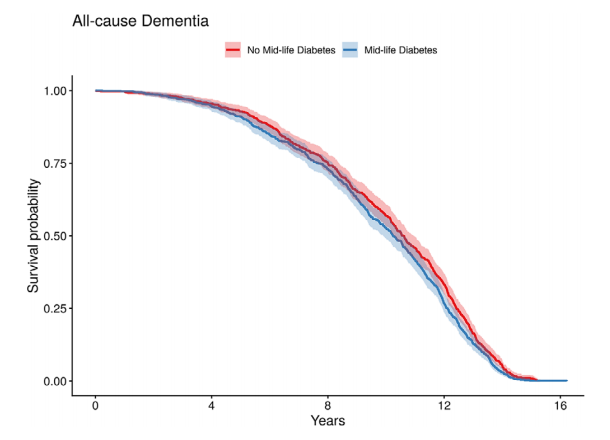
图 中年期2型糖尿病与痴呆发生风险
基于UK Biobank数据的Kaplan–Meier曲线显示,与对照人群相比,中年期诊断2型糖尿病的个体后续发生全因痴呆的风险更高。该结果提示,糖尿病对认知结局的影响可能与暴露年龄和病程长度有关,中年期代谢异常尤其值得关注。
同时,APOE基因背景也使问题更加复杂。APOE ε4是阿尔茨海默病最强的常见遗传风险因素之一,ε4/ε4携带者较APOE3/3人群风险显著升高。APOE4不仅与Aβ斑块、神经原纤维缠结、胶质反应和脑淀粉样血管病有关,也与脂质转运、免疫功能和血脑屏障完整性相关。糖尿病与APOE4之间并非简单叠加,但当2型糖尿病发生于APOE4携带者时,可能进一步放大阿尔茨海默病风险。
由此来看,糖尿病影响认知的路径并非单一路径,而可能同时包括血管损伤、代谢毒性、慢性炎症、脂质异常以及AD特异性神经病理改变。
小胶质细胞脂质滴,或是连接T2D与AD的新线索
近年来,单细胞和单核转录组学研究正在重新塑造人们对阿尔茨海默病病理机制的理解。与传统整体脑组织分析相比,单细胞研究能够进一步区分不同细胞类型和细胞亚型,尤其揭示了小胶质细胞在疾病不同阶段中的复杂作用。
小胶质细胞是脑内固有免疫细胞,既可以在早期参与Aβ清除和组织修复,也可能在疾病进展中转向促炎和促病理状态。TREM2–APOE信号通路是调节小胶质细胞从稳态向疾病相关状态转换的重要网络。多项研究显示,阿尔茨海默病中存在脂质相关小胶质细胞亚型,这些细胞具有胆固醇储存、脂质代谢和脂质分解相关基因表达特征。问题在于,当脂质负荷过重时,小胶质细胞可能形成过多脂质滴,吞噬Aβ的能力下降,同时更容易形成促炎性、疾病相关的细胞状态。
这一发现为2型糖尿病与阿尔茨海默病之间的联系提供了新的解释。2型糖尿病的核心异常并不只是高血糖,还包括外周脂质代谢紊乱、低密度脂蛋白胆固醇升高、脂毒性、慢性炎症、氧化应激、内质网应激、自噬异常等。上述改变可损伤脑血管内皮和血脑屏障,使外周炎症介质和免疫信号更容易影响脑内环境。血脑屏障“变漏”后,外周免疫信号、血管周围巨噬细胞与脑内小胶质细胞之间的交流可能被异常放大,推动小胶质细胞进入脂质异常和促炎状态。
从机制链条看,2型糖尿病可能通过“外周代谢异常—血脑屏障损伤—中枢免疫信号异常—小胶质细胞脂质代谢失衡—Aβ清除下降及tau病理促进”这一连续过程,增加阿尔茨海默病神经病理改变风险。这一过程可概括为炎症与脂质异常的交汇:高糖、高LDL胆固醇及毒性脂质促进外周和脑血管内皮炎症,血脑屏障通透性增加,疾病相关小胶质细胞形成脂质滴并降低Aβ吞噬,长期神经炎症环境进一步扰乱突触、髓鞘和神经元稳态,最终与Aβ斑块、tau异常磷酸化、神经原纤维缠结和神经元死亡共同推动认知下降。
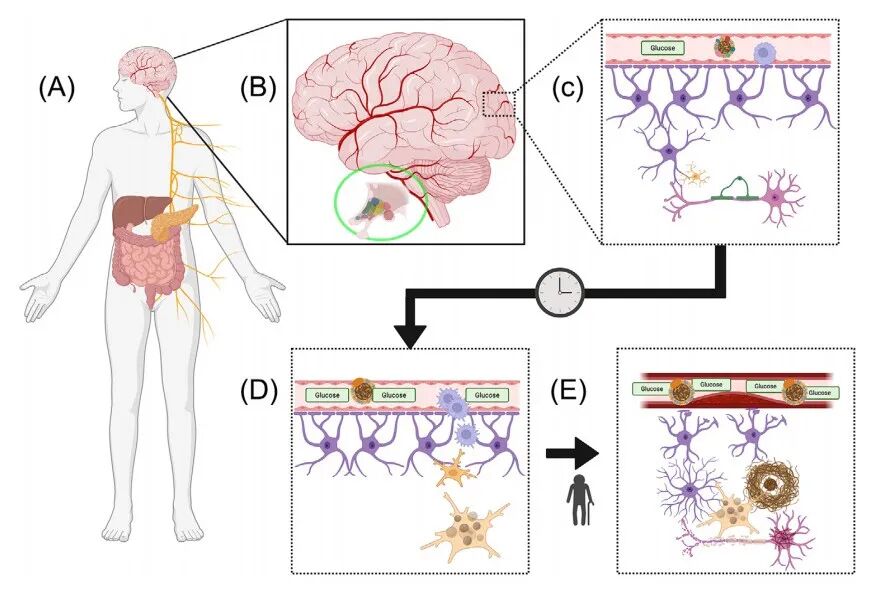
图 T2D促进AD发生的潜在通路。
(A)机体与大脑通过体液与神经通路双向连接,脑–肝–肠轴经迷走神经相连。迷走神经向内脏(包括肝脏、胰腺)发出传出纤维并接收传入信号。肠道菌群与酶协同分解碳水化合物、脂肪与蛋白质。西式饮食中过量的脂肪与糖类会增加肠道通透性并引发炎症。肠道同时释放饥饿素、GLP‑1 等肽类物质,调控食欲与胰岛素分泌。营养物质经门静脉到达肝脏,被储存或代谢。肠道来源的过量糖脂超出肝脏代谢能力,会引发脂肪蓄积与脂肪性肝炎。胰腺应对餐后高血糖分泌胰岛素,但慢性高血糖会导致肝脏、肌肉等胰岛素敏感器官发生胰岛素抵抗,最终造成 β 细胞功能衰退与 2 型糖尿病。(B)大脑通过迷走神经与血液监测肠道状态。大脑外周被血脑屏障(BBB)包裹,仅小分子物质可自由透过。葡萄糖经脑血管内皮细胞上的非胰岛素依赖性 GLUT1 受体转运入脑。大脑第三脑室周围存在室周器,该区域血脑屏障通透性更高,邻近神经元可感知血液与脑脊液中的营养物质、电解质、激素、趋化因子与细胞因子。这些神经元与内脏调节中枢(如下丘脑)及下丘脑‑垂体‑肾上腺轴(HPA)相连。HPA 在 2 型糖尿病中过度激活,进一步加重胰岛素抵抗并促进炎症。(C)生理状态下,葡萄糖顺浓度梯度被摄取。大脑重量仅占体重 2%,却消耗机体 70% 的葡萄糖与 20% 的氧气,其能量消耗与脑活动高度相关,该过程称为神经血管耦合。调控该过程的核心细胞为星形胶质细胞,其足突连接血管与突触。星形胶质细胞足突紧密连接,构成血脑屏障内层。脑动脉侧的两层血脑屏障结构间为血管周围间隙,巨噬细胞等免疫细胞在此与脑实质细胞相互作用。星形胶质细胞之间、与小胶质细胞、神经元及少突胶质细胞均可进行信号交流。小胶质细胞是大脑的巨噬细胞,持续监测周围微环境以维持脑功能与应对损伤。少突胶质细胞突起富含髓鞘脂质,包裹神经元形成髓鞘,显著提升动作电位传导速度。(D)2 型糖尿病以高血糖与低密度脂蛋白(LDL,“坏” 胆固醇)升高为特征。高血糖与高血脂引发广泛炎症,包括脑血管内皮炎症,导致血脑屏障通透性增加,炎症介质甚至免疫细胞更易进入脑实质。血管周围巨噬细胞可直接向小胶质细胞传递信号并诱导其活化。这些外周刺激促使小胶质细胞向疾病相关表型转化,表现为脂滴形成、对 Aβ 的吞噬能力下降。(E)长期 2 型糖尿病伴随的高血糖、LDL 胆固醇、神经酰胺等毒性脂质、炎症介质与免疫细胞浸润,会导致慢性外周炎症与血脑屏障渗漏,推动小胶质细胞向促炎表型转化。持续的神经炎症微环境扰乱神经元与胶质细胞功能,包括突触与髓鞘维持、Aβ 斑块清除。神经元启动应激反应,但长期功能失衡导致激酶与磷酸酶活性紊乱。tau 蛋白异常过度磷酸化后易自我聚集,在神经元胞体形成神经原纤维缠结(NFTs),最终导致神经元死亡。海马区等关键脑区神经元丢失引发 AD 早期
降糖药能否降低AD风险?
如果2型糖尿病确实参与阿尔茨海默病风险,那么降糖药物是否可能降低认知下降或AD风险,自然成为临床关注的问题。既往队列研究提示,在2型糖尿病患者中,SGLT2抑制剂、GLP-1受体激动剂和DPP-4抑制剂可能与较低AD风险相关,而
但从治疗研究看,证据仍需谨慎解读。针对早期症状性阿尔茨海默病的两项口服
这一结果反而强化了一个重要认识:控制代谢疾病对于整体健康和长期脑血管风险至关重要,但单纯纠正外周代谢异常,未必足以逆转已经形成的阿尔茨海默病病理过程。未来更有希望的方向,可能是基于具体机制进行分层干预,例如识别存在明显神经炎症、血脑屏障损伤或小胶质细胞脂质异常的高风险人群,再探索更精准的干预策略。
TREM2–APOE信号通路因此成为潜在治疗靶点之一。激活TREM2、增强小胶质细胞吞噬功能的单克隆抗体AL002已进入Ⅱ期研究,但在轻度
临床启示:认知风险管理需要更早看见代谢与炎症
对于神经科临床而言,我们并不应该把阿尔茨海默病重新命名为某种糖尿病,而是提醒我们:中年期长期存在的2型糖尿病、脂质异常、肥胖、血管损伤和慢性炎症,可能在症状出现前多年就已开始影响脑内微环境。
在认知障碍评估中,糖尿病史不应只被视为一般合并症。患者的发病年龄、病程长度、血糖控制情况、血脂状态、肥胖程度、脑血管病变、APOE遗传背景以及炎症相关生物标志物,未来都有可能共同帮助识别不同风险亚型。尤其值得关注的是,在糖尿病且超重或肥胖人群中,GFAP和NfL随时间升高可预测认知下降,而Aβ1-42/Aβ1-40比值或p-tau181未显示同样预测作用。这一发现提示,神经炎症或神经胶质损伤信号,可能比传统Aβ/tau标志物更早反映部分糖尿病相关认知风险。
从预防角度看,中年期代谢健康管理仍然具有重要意义。控制血糖、改善脂质异常、减少肥胖和血管风险因素,可能不仅是心脑血管病防控策略,也是认知障碍防控中的重要组成部分。但从治疗角度看,针对已经发生的阿尔茨海默病,仅靠降糖或减重可能远远不够。未来需要将代谢状态、血管因素、神经炎症、脑影像、血液生物标志物和遗传风险整合起来,才能更准确地识别哪一类患者可能从特定干预中获益。
结语
2型糖尿病与阿尔茨海默病之间的关系,正在从“中枢胰岛素抵抗”这一单一假说,走向更复杂也更接近临床现实的多机制模型。糖尿病不仅影响血糖,也影响脂质代谢、血管内皮、外周炎症和血脑屏障;这些改变可能进一步塑造脑内小胶质细胞状态,使其从保护性清除细胞转向脂质负荷过重、吞噬功能下降、促炎和促病理进展的状态。
这一认识有助于重新理解代谢病与神经退行性疾病的交叉地带。阿尔茨海默病不是简单的“脑内糖尿病”,2型糖尿病也不是只通过血管病变影响认知。真正值得关注的,或许是长期代谢紊乱如何悄然改变脑内免疫环境,并在多年后转化为认知功能下降的风险。对于未来的AD防治而言,代谢健康、血管健康与神经免疫稳态,可能需要被放在同一张长期管理图谱中共同审视。
参考文献:Sutherland GT, Chen A, Nguyen-Hao H-T, et al. How does type 2 diabetes modify the risk of Alzheimer’s disease? Alzheimer’s & Dementia. 2026;22:e71471.
 我要投稿
我要投稿















{kind=link}