

多年来,研究者提出了不同的模型来描述肿瘤的发生、进展和异质性。自19世纪始,肿瘤干细胞(CSC)的概念在科学界逐渐引起关注。组织学家Ernst Neumann和Artur Pappenheim在研究各种形式的
研究显示,肝癌干细胞(LCSC)呈现出以下独特的生物学特征:(1)高度致瘤性:在肿瘤组织中占比极少,却拥有远超普通肿瘤细胞数百倍的成瘤能力;(2)自我更新与分化潜能:具备类似正常干细胞的自我复制与分化潜能,可分化为恶性细胞、血管内皮细胞等多种肿瘤组分,促进肿瘤异质性形成,影响疾病进展、转移与复发;(3)持续增殖特性:凭借无限分裂能力驱动肿瘤恶性生长与治疗后再生;(4)治疗抵抗机制:对化疗、放疗等传统治疗表现出耐受和抗凋亡能力,残留细胞可重新增殖,致使癌症复发与耐药。
本研究系统剖析LCSC生物学特征,重点聚焦致瘤潜能、自我更新、多向分化潜能与持续增殖等特性,进一步深入研究LCSC内信号转导网络、微环境相互作用机制及表观遗传调控等,有望揭示其异质性形成的分子基础,有助于构建更精准的肿瘤治疗新模式,为创新临床干预策略提供理论支撑。

1LCSC的起源与演化
肝脏是一个兼具外分泌与内分泌功能的器官,并具有强大的再生能力。研究表明,正常肝细胞可在约1年内完全更新一次,这体现了干细胞的重要特征。在肝脏中,内源性干细胞(通常指位于末端胆管和小叶间胆管旁的肝卵圆细胞,也称肝前体细胞)数量庞大,但其增殖潜能的持续时间较短。这类卵圆细胞具有分化为肝细胞和胆管细胞的双向潜能,其表面标志物包括肝卵圆细胞标记6、细胞角蛋白7和19、甲胎蛋白、KIT原癌基因受体酪氨酸激酶和Thy-1细胞表面抗原等。在HCC中,被称为LCSC的细胞群体具有类似的干细胞特征并控制分级结构,它们是肝癌发生、发展、耐药和复发的关键细胞来源。在
针对HCC细胞起源的争论长期存在,一种理论认为其来源于去分化的成熟肝细胞。肝细胞去分化指成熟肝细胞在损伤或致癌压力下,逆转为类肝前体细胞的过程,表现为干性相关基因(如Sox9、Sox4、Cd24a)的重新激活及分化功能丧失。另有学者则认为,HCC细胞是从肝干细胞的异常分化中通过“成熟受阻”获得的,例如通过化学致癌物和致癌基因干预肝卵圆细胞的分化过程,使其转化为HCC癌前细胞。

2LCSC的生物学特性
LCSC具有分化成不同细胞谱系的能力,并能在移植至体内后,产生出一系列能够反映原发性肿瘤异质性的可移植肿瘤。醛脱氢酶1A1阳性LCSC在免疫缺陷小鼠中仅需100个细胞即可成瘤,而阴性细胞即使注入104个仍无致瘤性,二者差异达100倍;同样,CD24+LCSC仅需500个细胞即可启动肿瘤发生。临床队列分析进一步表明,CD24+高表达患者术后1年复发率增加3倍。
研究发现,LCSC所依赖的信号通路类似于早期胚胎发育期间调控细胞命运的信号通路[Notch、Wnt、Hedgehog和转化生长因子-β(TGF-β)]。癌症相关成纤维细胞(CAF)来源的外泌体中长链非编码RNA(lncRNA)NEAT1,可通过促进转录因子Yes相关蛋白的液-液相分离,进而激活HCC干细胞特性,揭示了一种新的调控机制。
研究表明,LCSC常处于G0期(静止期),依赖细胞周期抑制蛋白维持静息状态,从而避免进入活跃分裂阶段。近期研究发现,血管内皮生长因子-成纤维细胞生长因子(VEGF-FGF)信号通路可作为激活肝干细胞的分子开关,微环境变化可有效地调控其命运走向。而活化后的LCSC可通过关键通路实现无限增殖,如溴结构域PHD指状转录因子通过调控hTERT
通常认为,LCSC介导的治疗耐药性与

3LCSC在HCC进展过程中的作用
过去的30多年以来,对关键LCSC表面标志物的研究已取得显著进展。大量证据已阐明,CD133+细胞具有干细胞相关特征,包括放化疗抵抗、自我更新、致瘤性和转移等潜能。研究还发现,多种信号通路的内在激活参与调控CD133+LCSC的行为,如Wnt/β-catenin、蛋白激酶B、丝裂原激活的蛋白激酶/胞外信号调节激酶信号传导及TGF-β。除CD133之外,CD13也被确认为HCC中一种重要的干细胞表面标志物,可通过与组蛋白去乙酰化酶5相互作用阻止p65降解并激活NF-κB信号通路,从而促进HCC进展并诱导索拉非尼耐药。Wnt/β-catenin和IL-6/STAT3能够调控CD133、EpCAM、CD24等多种阳性标志物的LCSC干细胞特性,因此,靶向这两条通路被视为消除LCSC及逆转其耐药性的合理策略。
尽管不同阳性标志物的LCSC群体共享部分分子特征与调控通路,但它们仍具有独特的生物学特性。多项研究显示,特定基因表达模式与HCC患者临床预后相关,表明不同的LCSC亚群转录特征可能影响肿瘤异质性及疾病进展。对60例HCC患者手术标本的流式分析显示,LCSC标志物表达存在明显差异,其中CD24、CD13与EpCAM表达变异是异质性主因,约53.3%(32例)样本中这3种标志物共表达,且与肿瘤晚期进展显著相关,这为HCC分子分型及临床转归研究提供了新依据。
借助HCC动物模型以及基因阵列分析技术,诸多研究表明,众多已知在干细胞维持、自我更新及多能性调控中的信号通路在HCC中发生异常。这种改变可能导致LCSC的恶性转化。
Wnt信号通路异常见于约三分之一的HCC病例中,凸显其在癌症发生机制中的关键作用。当Wnt配体与卷曲受体结合时,通常会触发信号级联反应,如人源化抗体2-6可通过阻断EpCAM裂解,抑制其上皮细胞黏附分子胞内结构域的核转移,并下调Wnt受体表达,从而抑制肿瘤生长;γ-分泌酶抑制剂DAPT可阻止EpCAM剪切,减少上皮细胞黏附分子胞内结构域的释放;XAV939(β-catenin抑制剂)能促进β-catenin降解,进而降低EpCAM+LCSC的干细胞特征;ICG-001(β-catenin抑制剂)则通过阻断β-catenin与T细胞因子4的相互作用,抑制EpCAM的表达(图1)。卷曲受体随后向β-catenin发出信号,促使其脱离与E-钙黏蛋白的结合。研究表明,β-catenin具有双重作用:既能加速癌细胞增殖,又会削弱细胞间黏附,从而共同推动肿瘤发展,并常预示不良预后。另有研究发现,泛素特异性蛋白酶8通过β-catenin的翻译后修饰机制激活Wnt/β-catenin信号,进而促进HCC进展并抑制铁死亡。靶向泛素特异性蛋白酶8可能是HCC患者治疗的一种潜在策略。
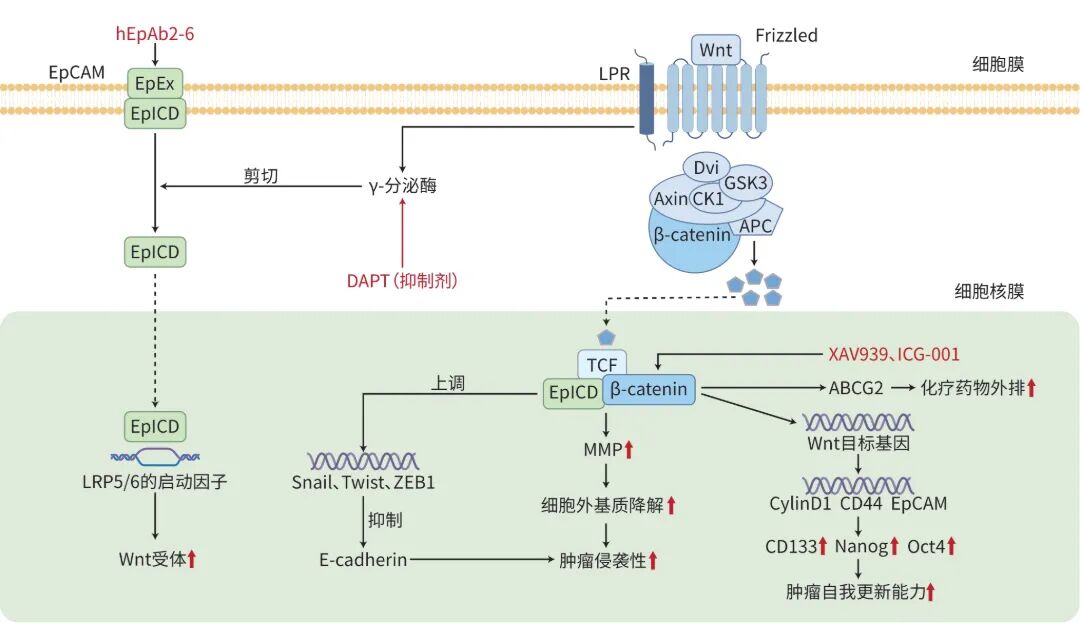
注: hEpAb2-6,人源化抗体2-6;EpCAM,上皮细胞黏附分子;EpEx,上皮细胞黏附分子胞外结构域;EpICD,上皮细胞黏附分子胞内结构域;DAPT,γ-分泌酶抑制剂;Frizzled,卷曲受体;LRP,低密度脂蛋白受体相关蛋白;Dvi,蓬乱蛋白;GSK3,糖原合成酶激酶3;CK1,
图1 LCSC表面的EpCAM通过Wnt/β-catenin信号通路发挥作用机制图
Hedgehog信号通路是由一系列分子构成的复杂体系,在干细胞生物学过程中发挥着调节作用。典型的Hedgehog信号通路主要由3种配体激活,这些配体能够与细胞膜上的2种12次跨膜蛋白受体——PTCH1和PTCH2发生相互作用。在缺失配体的环境下,作为一种7次跨膜的G蛋白偶联受体样信号转导分子,平滑受体(SMO)被局限于囊泡内而处于非活状性态。当配体与PTCH1或PTCH2结合,便会解除对SMO的抑制,促使其转移至细胞膜上的初级纤毛,而5E1抗体能够中和配体,从而有效阻断旁分泌信号途径;IPI-926可破坏CAF-LCSC信号轴;Vismodegib、Glasdegib则通过抑制SMO活性,抑制神经胶质瘤相关癌基因同源蛋白(Gli)核转位(图2)。该过程触发了关键转录因子胶质瘤相关癌基因1向细胞核内的迁移,进而激活一系列下游靶基因,包括胶质瘤相关癌基因1、PTCH1、细胞周期蛋白D、血管内皮生长因子以及c-myc等。一项多中心队列研究(n=287)显示,在HBV相关HCC患者中,Hedgehog信号通路激活亚型占34%,且该亚型对免疫检查点抑制剂的客观缓解率仅为8%,显著低于非激活亚型。Hedgehog基因的激活模式与HCC不良临床结局和TME异质性相关。从治疗角度看,Hedgehog信号通路阻断不仅可抑制HCC细胞增殖,还能提高肿瘤细胞对5-氟尿嘧啶的敏感性并诱导细胞凋亡。基于此,联合Hedgehog信号通路特异性抑制剂与传统5-氟尿嘧啶化疗,有望为HCC治疗开拓新路径,其临床应用潜力值得深入探究。
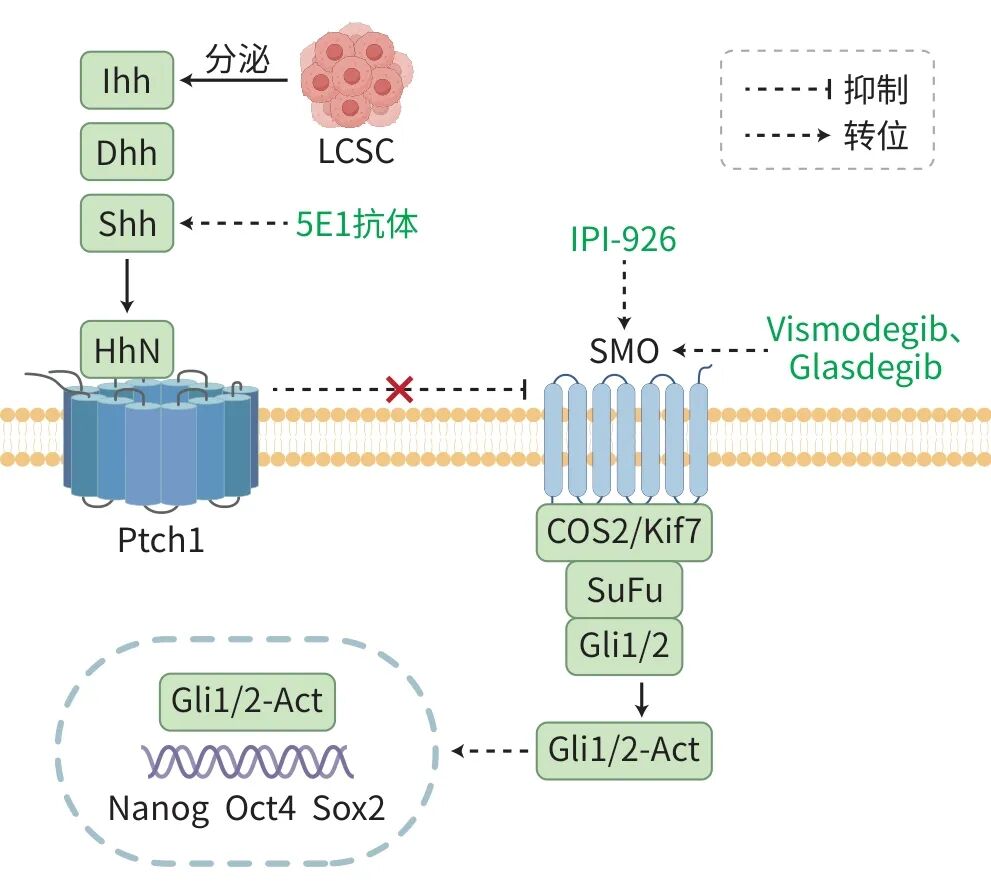
注: LCSC,肝癌干细胞;Ihh,印度刺猬蛋白;Dhh,沙漠刺猬蛋白;Shh,sonic刺猬蛋白;HhN,刺猬蛋白N端结构域;PTCH1,patched同源蛋白1;IPI-926,Hedgehog通路抑制剂;SMO,smoothened同源蛋白;COS2/Kif7,Costal-2/驱动蛋白家族成员7;SuFu,抑制融合蛋白;Gli1/2,Gli转录因子1/2;Gli1/2-Act,活化的Gli转录因子1/2;Nanog、Oct4,干细胞相关转录因子;Sox2,SRY框转录因子2;Vismodegib、Glasdegib,Hedgehog通路抑制剂;5E1抗体,抗sonic刺猬蛋白单克隆抗体。
图2 Hedgehog通路维持LCSC干细胞特性机制图
厦门大学一项研究显示,早期HCC细胞中糖原过度累积可形成相分离液滴,包裹Hippo通路激酶Mst1/2并抑制其活性,导致Yes相关蛋白去磷酸化并入核激活促干性基因。
研究显示,糖原累积是肝脏恶性转化过程中的关键致癌事件。癌症起始细胞适应糖原储存模式,并借助糖原相分离阻断Hippo信号通路,从而增加肿瘤发生风险。靶向CAF的同型靶向纳米粒子可结合维生素B3代谢重编程,减少CSC和抑制性免疫细胞群体,增强癌细胞对药物的敏感性,并促进杀伤性T细胞浸润,从而在小鼠HCC模型中恢复化疗敏感性。
有研究报道,在鉴定了140个在HCC中高度上调的lncRNA候选结合蛋白后,通过相互作用组分析发现,lncRNA HULC能够通过调控乳酸脱氢酶A和M2型丙酮酸激酶促进有氧糖酵解。微RNA(miRNA)-122作为肝脏特异性miRNA,其缺失导致M2型丙酮酸激酶和乳酸脱氢酶A表达上调,促进糖酵解并维持LCSC的干细胞特征;而恢复miRNA-122表达可抑制肿瘤生长。
近年来的研究越来越强调TME在耐药性发展及恶性进展中的关键作用。TME由血管网络、免疫组分、细胞外基质细胞、信号传递因子等共同构成。
髓系细胞重编程在LCSC免疫调控中具有重要作用。单细胞转录组分析表明,LCSC通过分泌信号素3C、CC趋化因子配体2、CC趋化因子配体5等,主动募集并调控髓系来源抑制细胞和肿瘤相关巨噬细胞的功能状态。南京医科大学团队于2024年在Signal Transduction and Targeted Therapy发表的研究进一步揭示,信号素3C通过激活肝星状细胞中的NF-κB信号通路,刺激IL-6释放并上调3-羟基-3-甲基戊二酰辅酶A还原酶的表达,从而促进胆固醇合成;这些胆固醇可被LCSC摄取,以增强免疫检查点分子的膜定位和信号转导效率。此外,CAF分泌的TGF-β1通过激活HCC细胞中的AP1信号,上调信号素3C表达,形成正反馈环路,加速HCC进展。值得注意的是,阻断信号素3C不仅能有效抑制肿瘤生长,还可提高HCC细胞在体内对索拉非尼的敏感性。
LCSC具有独特的免疫调节能力,能够主动重塑肿瘤免疫微环境,形成有利于自身存活与增殖的免疫抑制性生态位。该过程涉及多种免疫细胞的表型调控与功能改变,是LCSC实现免疫逃逸的关键机制。一项针对11例初治HBV相关HCC患者(包括8例细胞角蛋白19阳性)活细胞的单细胞RNA测序研究发现,血管内皮生长因子A信号可激活分泌性磷蛋白1阳性肿瘤相关巨噬细胞衍生的基质金属蛋白酶9,进而促进细胞角蛋白19阳性HCC肿瘤的侵袭与转移。空间转录组技术的引入弥补了单细胞RNA测序在空间信息保留方面的缺陷。2022年,清华大学团队在第八届全国计算生物与生物信息学大会上报告,利用10X Genomics平台对7例HCC患者的21个组织样本进行空间转录组分析,共获得84 823个空间测序位点数据,成功绘制了HCC的高分辨率空间图谱,并开发出TLS-50标记以精确定位三级淋巴结构。2024年9月20日,Nature Cancer杂志上发表的一项研究显示,斯坦福大学团队利用CO-Detection by indexing技术分析100多例人类HCC样本,精确描绘了治疗后残留LCSC与M2样巨噬细胞形成的“免疫豁免微区”的空间特征。研究发现,表达程序性死亡配体1(PD-L1)的巨噬细胞通过与癌细胞直接接触,激活其TGF-β信号,导致T细胞功能耗竭,从而促进免疫逃逸与肿瘤复发。该研究进一步揭示了PD-L1+巨噬细胞可通过激活干细胞样癌细胞中的TGF-β维持多药耐药状态,而靶向该相互作用有望预防多药耐药的HCC复发。
免疫检查点分子的协同表达是LCSC实现免疫逃逸的核心策略。除PD-L1外,LCSC还高表达CD47、人类白细胞抗原G等分子,可向巨噬细胞和自然杀伤细胞传递“别吃我”信号。2024年加州大学圣地亚哥分校在Cell Stem Cell发表的研究显示,阻断TGF-β信号可显著增强诱导多能干细胞来源的自然杀伤细胞对LCSC的杀伤效果,证实TGF-β是LCSC免疫逃逸的关键介质。该团队通过基因工程使自然杀伤细胞中TGF-β受体失活,结果发现这些细胞对HCC的清除能力大幅提升,并有效延长小鼠生存期。研究者强调,双重免疫阻断策略(如同时靶向PD-L1和TGF-β)可能是克服LCSC介导的免疫抑制的更有效方案。
此外,CSC可通过上调PD-L1、PD-1、细胞毒性T细胞相关蛋白4等免疫检查点分子模拟免疫细胞表型,从而抑制T细胞活性并逃避免疫系统的监视,这一现象被称为“免疫细胞模拟”。CSC还可通过下调主要组织相容性复合体Ⅰ类分子表达,逃避CD8+T细胞的识别。

4LCSC靶向治疗新策略
嵌合抗原受体T细胞免疫疗法(CAR-T)通过基因工程改造患者自身T细胞,使其表达可特异性识别肿瘤表面抗原的合成受体,从而实现对肿瘤细胞的精准清除。近年来,CAR-T疗法在靶向LCSC方面取得重要进展,其作用机制主要依赖于识别LCSC特异性或高表达的抗原表位。科济药业开发的CTO11是全球首个针对磷脂酰肌醇蛋白聚糖-3的CAR-T疗法,在用于晚期HCC的Ⅰ/Ⅲ期临床试验中,已有2例患者实现9年的无癌生存,显示出该疗法在长期控制LCSC方面的潜力。上海雅吉生物等机构研发的针对EpCAM和CD133的双靶点CAR-T,在临床前研究中显示出对LCSC的特异性清除能力。
格拉吉布联合PD-1抗体(临床试验号NCT04035876)可显著延长患者的中位无进展生存期。SMO蛋白抑制可使CD133+LCSC比例达60%。白三烯A4水解酶通过靶向异质核核糖核蛋白A1/潜在转化生长因子β结合蛋白1/TGF-β信号轴,改善TME并抑制HCC进展。
2-脱氧葡萄糖可抑制LCSC的糖酵解过程,减轻免疫抑制微环境;联用PD-1抗体(如帕博利珠单抗)可显著提升肿瘤内CD8+T细胞浸润水平(提高2~3倍)。在HCC患者来源的肿瘤异种移植模型中,该联合方案使肿瘤体积缩小60%,并降低CD133+LCSC比例。线粒体复合物Ⅰ抑制剂则可选择性抑制LCSC的氧化磷酸化,诱导其凋亡(凋亡率增加80%),并下调CD133+LCSC干细胞相关标志物(如Nanog、Oct4)的表达。该抑制剂与抗细胞毒性T细胞相关抗原4联合治疗后,可显著增加肿瘤内CD8+T细胞浸润(较单药提高2倍)。在肝癌小鼠模型中,联合组肿瘤体积缩小70%,且中位生存期由14周延长至22周。
Nature Reviews Gastroenterology & Hepatology发表的一项研究显示,Galunisertib(TGF-β抑制剂)可抑制TGF-β信号通路,使CD133+LCSC比例显著降低(降幅达50%以上);联合PD-1抗体后,肿瘤生长抑制率提高80%,肺转移亦明显减少。其作用机制涉及免疫微环境的重塑,使“冷肿瘤”转化为“热肿瘤”。2024年在Nature Cancer杂志上一项重要研究进一步鉴定出POSTN1+2细胞外基质相关成纤维细胞作为关键的癌症-胚胎相互作用枢纽,促进肿瘤进展。细胞间通讯与空间转录组学分析揭示了癌症-胚胎细胞的交叉对话和共定位关系,包括POSTN3+4 CAF、FOLR25+6巨噬细胞及PLVAP7+8内皮细胞等群体。

5小结
综上,当前科研探索的新型干预策略主要聚焦于4个方面:靶向LCSC特异性抗原的免疫治疗、信号通路抑制剂联合免疫治疗、代谢干预新路径以及微环境重编程策略。通过靶向干细胞表面标志物,抑制Wnt/β-catenin、Notch、Hedgehog等信号通路活性,可干扰LCSC的增殖与分化;多种通路抑制剂在临床前研究中已表现出良好的抗肿瘤活性。在克服由ABC transporter家族介导的药物外排耐药机制方面,现有抑制剂普遍存在选择性差、毒性大等问题,因此,开发高效低毒的阻断剂仍是研究热点,优化药物设计与提高靶向特异性为未来方向。策略的实施需依据肿瘤的分子特征进行精准选择,多靶点联合干预有望突破现有治疗瓶颈。同时,针对不同肿瘤的特异性耐药机制,需开发相应的针对性治疗方案。
LCSC与TME的相互作用在肿瘤进展中发挥关键作用。微环境不仅调控干细胞的状态与功能,还为其在恶劣条件下的存活提供保护,探究该相互作用对开发新型治疗策略意义重大。LCSC的研究为理解癌症复杂性提供了新视角,深入剖析其生物学特性及作用机制,将有助于开发针对性治疗策略,从而提高患者5年生存率与生活质量。然而,当前研究仍存在精准识别靶向LCSC、干扰其自我更新分化机制等难题亟待解决。

https://www.lcgdbzz.org/cn/article/doi/10.12449/JCH260229

崔文婷, 刘宁宁, 马秀珍, 等. 肝癌干细胞在肝细胞癌中的作用和靶向治疗策略[J]. 临床肝胆病杂志, 2026, 42(2): 457-463
来源:临床肝胆病杂志
本平台旨在为医疗卫生专业人士传递更多医学信息。本平台发布的内容,不能以任何方式取代专业的医疗指导,也不应被视为诊疗建议。如该等信息被用于了解医学信息以外的目的,本平台不承担相关责任。本平台对发布的内容,并不代表同意其描述和观点。若涉及版权问题,烦请权利人与我们联系,我们将尽快处理。
 我要投稿
我要投稿















{kind=link}