
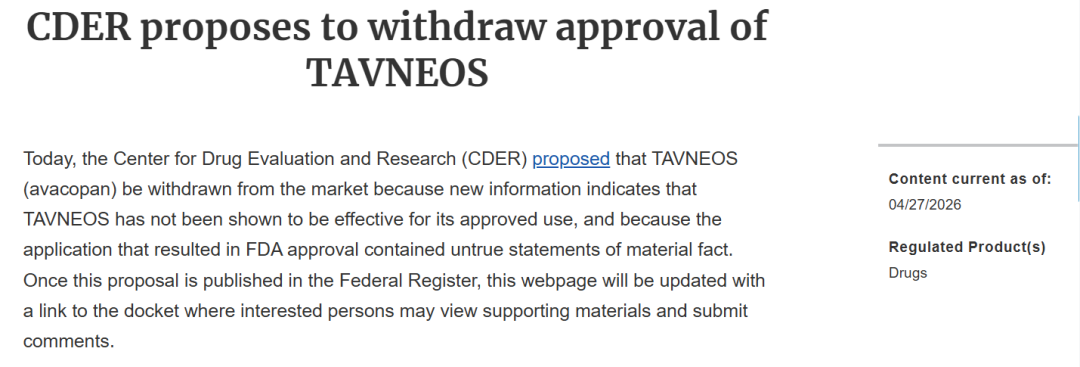
2026年4月27日,美国食品药品监督管理局(FDA)药物评价与研究中心(CDER)提出将

临床试验数据完整性存疑
FDA在2026年4月的撤市提案中披露,阿伐可泮的Ⅲ期ADVOCATE临床试验存在数据造假行为。该试验的关键终点为“第52周持续缓解率”,要求药物组疗效优于对照组且差异具有统计学显著性。2019年11月5日,该试验数据库首次锁定并揭盲,初步分析结果显示,原始p值为0.1025,未达到统计学显著标准,不符合上市审批要求。
经FDA核查内部记录发现,当时ChemoCentryx的首席医学官与生物统计学总监在知晓分组结果的情况下,选取了9名处于疗效判定边界的受试者,这些受试者的伯明翰
上市后严重安全性风险凸显
除数据造假问题外,阿伐可泮上市后出现的严重肝损伤案例也成为FDA提议撤市的重要原因。2026年3月31日,FDA发布药物安全通报,提示阿伐可泮与严重
FDA经核查发现,截至2024年10月9日,全球范围内已确认76例与阿伐可泮使用存在合理因果关系的药物性肝损伤病例,其中74例为严重病例,包括54例住院病例和8例死亡病例。在60例提供实验室信息的病例中,38例表现为
FDA表示,尽管肝毒性在阿伐可泮上市前的临床试验中已被识别并纳入产品标签,但上市后出现的胆管消失综合征及致命性肝损伤病例,构成了新的安全隐患。结合数据造假导致的疗效存疑问题,FDA认为该药物的获益已无法抵消其潜在风险。
事件进展与各方回应
2026年1月,FDA曾向安进发出函件,建议其自愿将阿伐可泮撤出美国市场,但安进在12天后回应称,未发现临床试验基础患者数据存在问题,且结合多年真实世界证据,有信心证明该药物的疗效及良好的获益-风险比,暂不打算撤市。
在FDA于4月27日启动行政撤市程序后,安进迅速作出回应,于4月29日向FDA提交了阿伐可泮的标签更新申请,主动加入胆管消失综合征的相关警告,此举被认为是安进试图通过承认安全风险,削弱FDA关于“安全性失控”的指控。
根据相关程序,安进可在2026年6月1日前向FDA申请听证会,为自身进行辩护。若FDA认为安进提交的辩护材料不足以构成“真实且实质性的重大事实争议”,可直接撤销阿伐可泮的上市批准,无需召开听证会。目前,该事件仍处于等待FDA进一步裁定的阶段。
此外,安进目前还面临来自欧洲和中国监管部门的相关调查,阿伐可泮在这些地区的市场前景也将取决于各地监管机构的后续评估结果。
相关背景补充
ANCA相关性血管炎是一组罕见的多系统自身免疫病,全球年发病率约为每百万人口10-20人,该病此前长期依赖糖皮质激素联合
参考来源
医脉通是专业的在线医生平台,“感知世界医学
 我要投稿
我要投稿















{kind=link}