近年来,代谢紊乱在ALI/ARDS发病中的作用日益受到重视。临床观察显示患者血浆中Omega-3多不饱和脂肪酸(PUFAs)水平显著下降,而脂肪酸去饱和酶FADS1/2作为PUFA合成的关键限速酶,其在ALI中的表达与功能变化尚不明确。与此同时,铁死亡(Ferroptosis)——一种由脂质过氧化和还原型谷胱甘肽(GSH)耗竭驱动的新型细胞死亡方式,与肺内皮损伤密切相关。由于PUFAs兼具细胞膜组分和脂质过氧化底物的双重角色,阐明FADS1/2-PUFA代谢轴在铁死亡中的调控作用至关重要。
在机制层面,抗氧化蛋白PARK7(DJ-1)在ALI中表现出保护作用,且在癌症等疾病模型中被证实能够抑制铁死亡。然而,PARK7是否通过调控FADS1/2影响PUFA代谢来调节铁死亡进程,尚待深入研究。此外,缺氧炎症状态下堆积的乳酸可作为表观遗传修饰(乳酸化)的底物,连接代谢与基因表达。但乳酸化修饰是否参与调控PARK7表达,进而影响FADS1/2-PUFA代谢与铁死亡,这一完整的调控轴尚属未知。
基于此,

1. 现象发现:“PUFA代谢-Ferroptosis”轴是内皮损伤的核心
➤在ALI状态下,肺内皮细胞的Omega-3 PUFAs合成通路被显著抑制,关键限速酶FADS1/2在转录及蛋白水平均明显下调,下游代谢产物
➤这一代谢紊乱直接导致脂质过氧化水平升高、
2. 中游解析:PARK7是代谢重整的关键开关
➤研究发现,抗氧化蛋白PARK7在ALI动物模型肺组织及患者血浆中均呈现浓度依赖性升高,提示其可能参与机体代偿性保护反应。通过构建PARK7过表达及干扰模型,进一步证实PARK7作为FADS1/2的上游内源性调控因子,直接参与PUFA代谢通路的调控。
➤PARK7通过靶向BMP-BMPR-SMAD1/5/9信号级联的双通路机制调节FADS1/2转录:一方面通过稳定转录因子NRF2促进BMP信号通路配体(BMP2/4)的合成;另一方面通过清除活性氧(ROS)维持RNA结合蛋白HuR功能,从而增强BMP受体(BMPR1A/B)mRNA稳定性。这两条通路的协同作用最终驱动p-SMAD1/5/9转录复合体入核,直接结合于FADS1与FADS2基因启动子区域,激活其转录表达,实现PUFA代谢重建与铁死亡的有效抑制。
3. 上游追溯:乳酸化修饰是回路的总触发器
➤ALI状态下,缺氧与炎症导致的乳酸大量堆积引发组蛋白表观遗传修饰的重编程,其中H3K14位点乳酸化(H3K14la)在肺内皮细胞中呈现特异性显著升高。H3K14la直接富集于PARK7基因的启动子区域,从而驱动其转录上调。
➤FADS1/2的功能缺失会进一步促进H3K14la修饰及PARK7转录。这表明,“H3K14la→PARK7 →FADS1/2”构成了一个自我强化的保护性反馈回路:。该回路在PUFA合成通路受损时被迅速激活,通过增强上游信号试图恢复代谢稳态,体现了细胞在应激状态下的一种高效自适应保护机制。
4. 通路验证:靶向该轴心展现治疗潜力
➤通过腺相关病毒(AAV)在体特异性过表达证实,仅在小鼠肺内皮细胞中过表达Fads1/2,就足以显著减轻脂多糖(LPS)诱导的ALI,包括缓解
➤构建全肺Fads1/2过表达模型并联合Omega-3前体(α-亚麻酸, ALA)补充,展现了协同增效的治疗作用,能更全面地改善各项肺损伤指标,并纠正脂质代谢失衡,为临床转化提供了前瞻性的实验依据。
该研究系统阐明了ALI中存在的“乳酸化-PARK7-FADS1/2-PUFA-铁死亡”调控轴。具体而言,肺内皮细胞在损伤状态下通过H3K14乳酸化修饰上调PARK7表达,进而通过BMP-SMAD1/5/9信号通路激活FADS1/2的转录,最终恢复PUFA代谢并抑制铁死亡过程。值得注意的是,研究发现当FADS1/2功能受损时,会进一步促进H3K14乳酸化与PARK7的表达上调,形成独特的代偿性放大机制。这一发现揭示,急性肺损伤中的内皮功能障碍在机制上与去饱和酶介导的脂质代谢紊乱、铁死亡性细胞死亡和乳酸依赖性的组蛋白修饰相关联,而PARK7是整合这些通路的关键节点(图1),这为理解肺内皮屏障功能障碍的分子基础提供了全新视角。
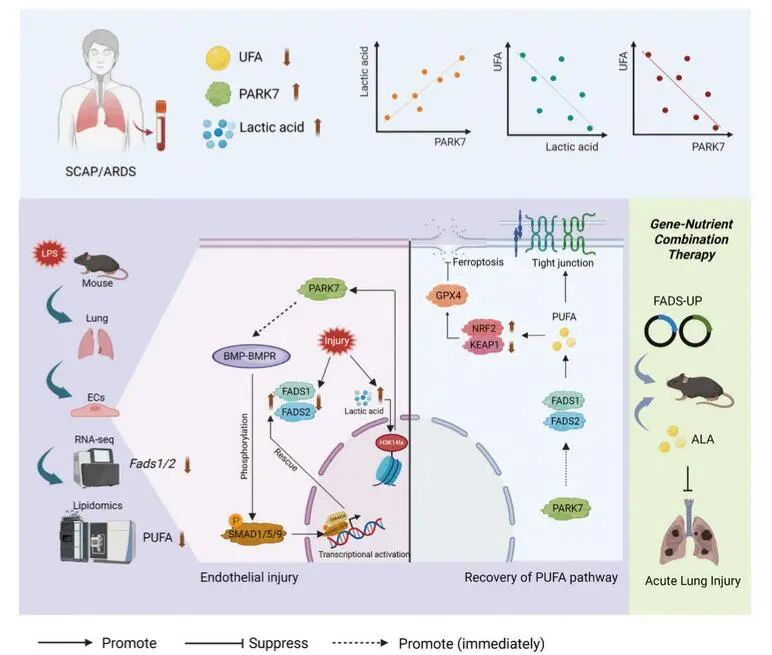
图1. 核心发现与机制假说
该研究的发现为ALI的防治开辟了多个富有前景的研究方向。在治疗策略层面,未来可致力于开发特异性PARK7激动剂、乳酸化修饰调控剂,并优化Omega-3脂肪酸的联合治疗方案。在转化研究方面,需要在更复杂的疾病模型中验证针对此通路的治疗效果,并探索该机制在其他器官损伤中的普适性。在机制探索层面,亟待解析FADS1/2初始下调的上游信号事件,阐明不同PUFA种类在铁死亡中的特异性作用,以及探索该通路与其他细胞死亡方式的交互网络。这些研究方向的推进将有助于将该基础研究发现转化为有效的临床治疗策略。
参考文献:Xu J, Wang Y, Mao W, et al. Metabolic Interplay in Acute Lung Injury: PARK7 Integrates FADS1/2-Dependent PUFA Metabolism and H3K14 Lactylation to Attenuate Endothelial Ferroptosis and Dysfunction. Adv Sci (Weinh). Published online September 30, 2025. doi:10.1002/advs.202508725

 我要投稿
我要投稿















{kind=link}