导语
近日,国家卫生健康委对《第二批罕见病目录》的86个病种分别制定了诊疗指南。 本文对涉及的肝病科五种罕见病(阿拉杰里综合征、
阿拉杰里综合征
阿拉杰里综合征(Alagille syndrome , ALGS)是一种常
临床诊断的确立依赖于综合判断。 经典的诊断标准为肝组织活检有肝内小叶间胆管数量减少或缺如,并具有至少包括慢性胆汁淤积、心脏杂音、蝴蝶椎骨、角膜后胚胎环和特殊面容等 5个主要临床表现的其中3个,并排除其他可能原因。 现在肾脏异常也列为主要异常之一。 如果肝活检不表现为肝内小叶间胆管数量减少或缺如,或由于某些成年轻症病人并未进行肝活检,修订的诊断标准认为符合4个或以上主要标准也可诊断。 如果已知有JAG1/NOTCH2 基因突变或阳性家族史时,2个主要标准通常即可确诊。
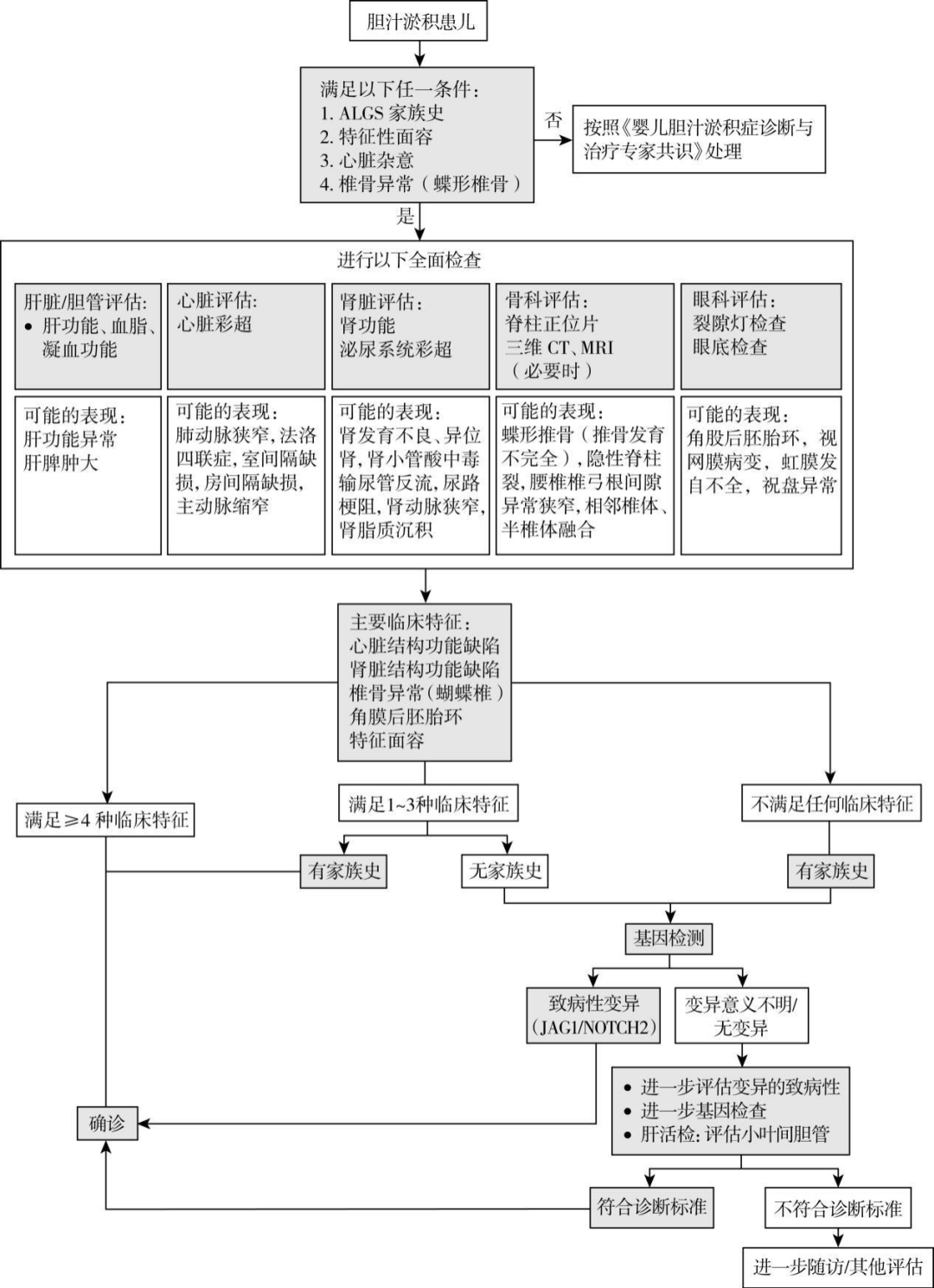
图1 阿拉杰里综合征诊疗流程
目前尚无特异的治疗手段,主要是支持治疗和对症处理。 由于该病有多系统受累,因此良好的管理需要多学科专科医师参与。 肝病方面主要面临的是胆汁淤积及其并发症,包括
ALGS 病人需要定期监测肝功能变化,必要时
α1-抗胰蛋白酶缺乏症
α1-抗胰蛋白酶缺乏症(a lpha1-antitrypsin deficiency,AATD)是一种由SERPINA1基因突变导致的常染色体共显性遗传疾病,可引起新生儿期
AATD 的诊断需结合慢性肝病和 COPD 的临床表现及相关辅助检查异常,血清AAT水平、AAT表型及基因型来综合判断。 诊断 AATD 的 AAT 水平的最佳界值为 1.1g/L,其区分正常基因型PI*MM与至少携带一个缺陷等位基因PI*MS或PI*MZ的灵敏度、特异度分别为73.4%、88.5%,但不能仅凭血清AAT水平来确诊或完全除外AATD。 AAT表型分析或基因型分析是诊断AATD的金标准,而肝活检 PAS-D 染色发现肝细胞内包涵体是诊断AATD所致肝病的金标准。
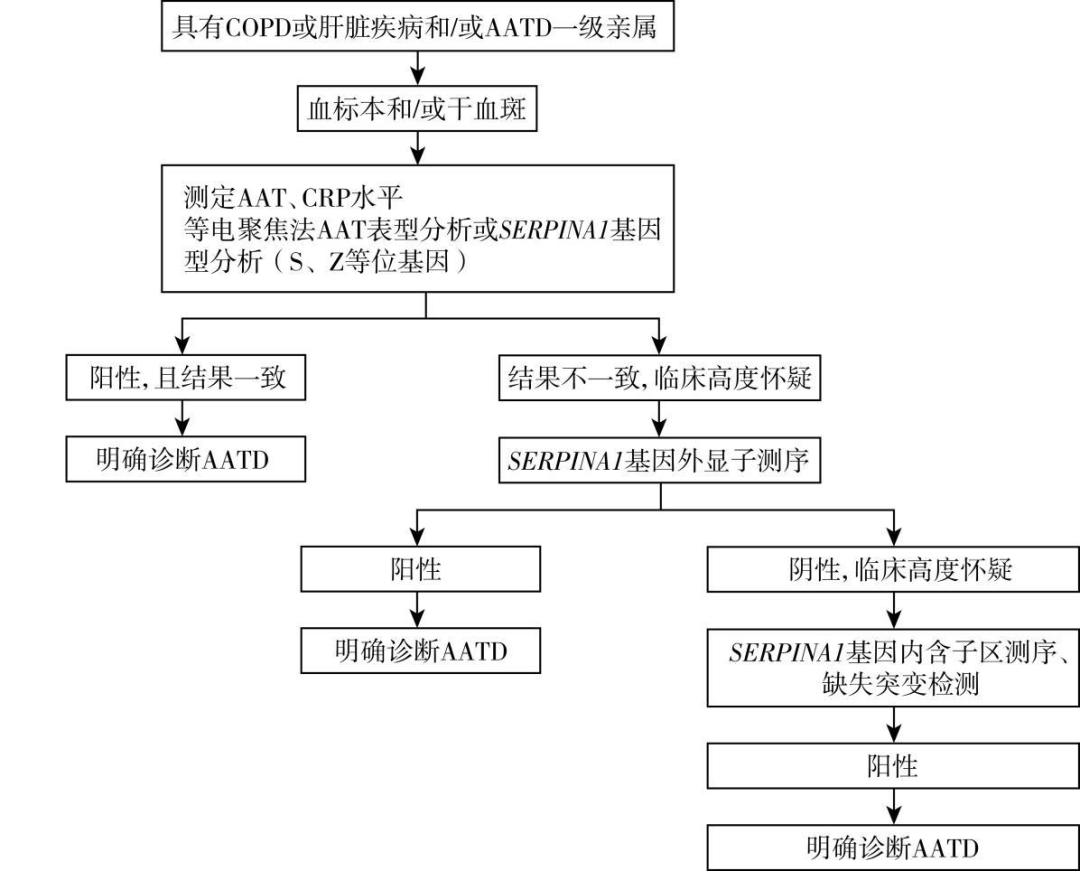
图2 α1-抗胰蛋白酶缺乏症的实验室诊断流程
(1)一般治疗:主要采取支持性措施,以预防或减少慢性肝脏疾病并发症。 戒酒、降低体重、控制糖尿病等,以延缓肝脏疾病进展。 如已发生肝硬化、
(2)肝移植:对于进入终末期肝病的AATD患者,肝移植是唯一有效的治疗手段。 对于AATD所致终末期肝脏疾病成人患者,肝移植的5年生存率为 85%。 有研究显示,采用PI*MZ杂合突变的供体也有很好的安全性和临床疗效。
(3)新兴治疗手段:目前正在研发多种针对AATD肝病的基因或细胞治疗手段,以抑制突变AAT的表达,促进AAT的分泌、降解或自噬。 其中,如基于小干扰RNA技术的fazirsiran 多中心开放 2 期临床试验显示,对 PI*ZZ AATD
先天性胆道闭锁
先天性胆道闭锁(congenital biliary atresia,CBA)是婴儿时期常见的严重肝胆系统疾病之一,以肝内、外胆管进行性炎症和纤维化为特征,早期表现为新生儿胆汁淤积症,晚期会出现胆汁性肝硬化、门静脉高压、
先天性胆道闭锁的早期筛查至关重要,比较简单易行的筛查办法是通过新生儿常规行大便比色卡筛查胆道闭锁。 目前日本、加拿大等国家和我国台湾地区已广泛开展粪便比色卡筛查,可有效降低胆道闭锁患儿Kasai手术日龄,提高自体肝生存率。 此外,也有探索新生儿出生后行足跟血质谱检测胆红素进行初筛,阳性病例在 2 周后复测血胆红素以筛查胆道闭锁。
先天性胆道闭锁其他比较常用的辅助检查方法包括:①超声检查,胆囊形态不规则,小胆囊,胆囊不可见、肝门纤维块、进食后胆囊无收缩等对胆道闭锁有提示作用; ②放射性核素
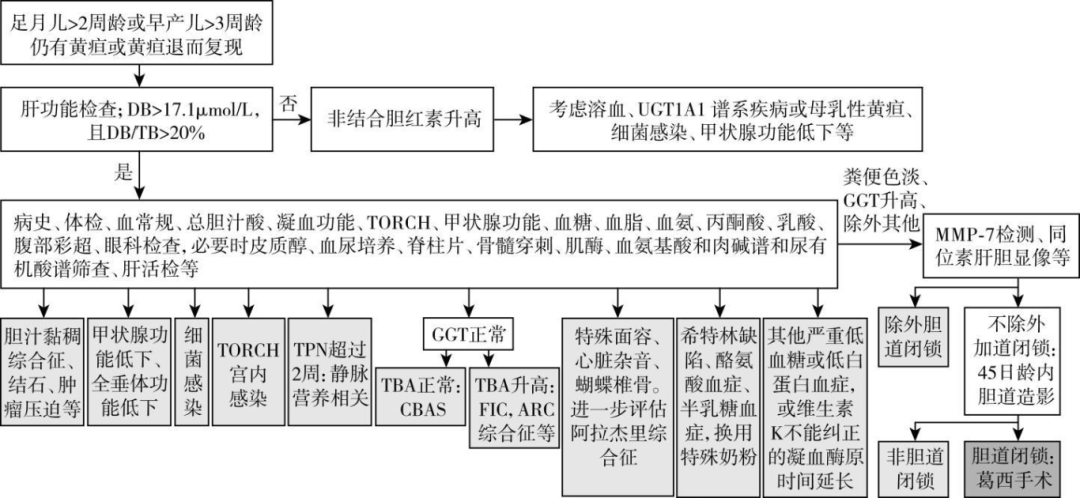
图3 先天性胆道闭锁和其他婴儿胆汁淤积诊断流程
➤手术治疗:胆道闭锁最重要的治疗手段是Kasai术。 成功的Kasai术可以恢复正常胆汁流,改善肝功能,以期达到长期存活的目的。
➤营养和支持治疗:胆道闭锁患儿的能量需求可达正常婴儿的 130%~150%,因此必须提供足量的营养支持,包括夜间加餐。 胆道闭锁患儿因胆汁分泌减少,造成其对食物中脂肪的消化和吸收能力减弱,对于胆红素水平升高的患儿,推荐使用富含中链甘油三酯(MCT)的奶粉喂养,或在食物中添加MCT。 胆汁淤积时由于胆盐
➤药物治疗:虽然Kasai术可以延长先天性胆道闭锁患儿的生命,但是不能逆转肝脏的损伤以及进行性肝硬化,应用药物治疗可以延缓肝硬化的发生时间。 术后通常需用熊去氧胆酸每日两次,每次每千克体重10mg口服来改善胆汁的排放情况。
原发性胆汁性胆管炎(primary biliary cholangitis,PBC,旧称
PBC的诊断需依据生物化学、免疫学、影像学及组织学检查进行综合评估。 满足以下3条标准中的2条即可诊断:(1)存在胆汁淤积的生物化学证据(主要是ALP和GGT升高),且影像学检查排除了肝外或肝内大胆管梗阻; (2)AMA/AMA-M2 阳性,或其他PBC特异性自身抗体(即抗-gp210、抗-sp100)阳性; (3)组织学上有非化脓性破坏性胆管炎和小胆管破坏的证据。

图4 原发性胆汁性胆管炎诊疗流程
➤一线治疗:熊去氧胆酸(UDCA)是治疗 PBC 的一线药物。 患者应长期口服 UDCA 13~15 mg/(kg·d)治疗,分次或一次服用。 治疗过程中需动态监测体质量变化,及时调整UDCA剂量。 UDCA安全性良好。 其不良反应较少,主要包括
➤治疗应答标准:国际上有多种评价UDCA治疗后生物化学应答的标准。 其中,巴黎Ⅰ和巴黎Ⅱ标准应用较多,分别用于评估晚期PBC 和早期PBC 患者生化应答。 此外,在新药临床试验中,多采用ALP≥1.67×ULN作为生化应答不佳的重要标准。 对于UDCA生化应答不佳的患者长期预后差、生存率低,需考虑二线治疗。
➤二线治疗:主要包括奥贝胆酸(obeticholicacid,OCA)、贝特类药物等。 对UDCA生化应答不佳的患者,可加用OCA进行联合治疗,剂量为 5~10 mg/d。 对于目前或既往有肝硬化失代偿事件(腹水、肝性脑病、食管胃静脉曲张破裂出血)、凝血功能异常及持续性血小板减少者,禁用OCA。 代偿期肝硬化患者使用OCA,需严密监测疾病变化。 对UDCA 应答不佳的患者,也可联合
原发性硬化性胆管炎(primary sclerosing cholangitis, PSC)是一类以肝内外胆管弥漫性炎症和纤维化(纤维-闭塞性胆管病)为特征的自身免疫性肝病,患者常合并
目前尚无国际上公认的PSC诊断标准,由于缺乏特异性的血清学及影像学诊断标志物,PSC诊断需结合患者临床表现、生化、影像学、组织学检查等多种证据,并排除继发性的胆道损伤因素进行综合诊断。 根据2021年中华医学会肝病学分会发布的《原发性硬化性胆管炎诊断及治疗指南》,大胆管型 PSC 诊断标准为:
(1)胆管影像学检查具备 PSC 典型特征。
(2)以下标准至少满足一条:①胆汁淤积的临床表现及生化学改变(成人AKP升高、儿童GGT升高); ②合并 IBD 的临床或组织学证据; ③典型PSC肝脏组织学改变; (3)排除其他病因引起的继发性硬化性胆管炎。 对于胆道成像无PSC典型表现的,如果满足以上标准第2条的2条以上或仅有PSC典型胆道影像学特征可疑诊 PSC。
小胆管型 PSC 诊断标准为:
(1)近期胆道影像学无明显异常改变。
(2)典型 PSC 肝脏组织病理学改变。
(3)排除其他因素所致胆汁淤积。 如果患者胆道影像学无异常,肝脏组织学具有PSC特点但不典型时,若患者同时存在 IBD 临床或组织学证据以及胆汁淤积的生化改变时,也可诊断小胆管型PSC。
由于国际上目前并无制定关于PSC/AIH重叠综合征的指南规范,临床实践中广泛采用的是多数欧洲和美国研究中心报道的PSC/AIH诊断标准,即符合前述 PSC诊断标准的同时,满足国际自身免疫性肝炎小组(IAIHG)的AIH诊断的简化标准。
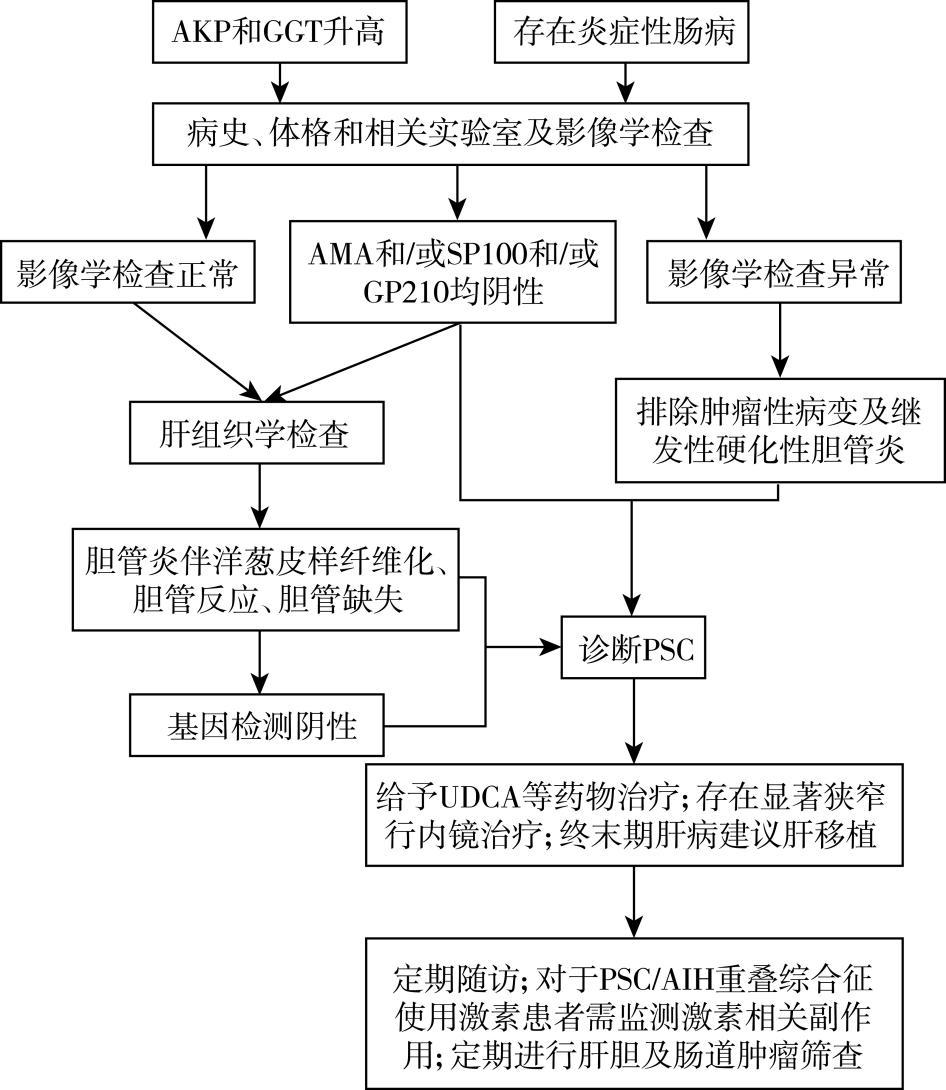
图5 原发性硬化性胆管炎诊疗流程
目前为止,国际PSC诊疗指南仍未明确推荐药物治疗方案。 目前有多种药物试用于PSC患者,包括熊去氧胆酸(UDCA)、抗生素、免疫抑制剂,以及调脂药、益生菌等。 内镜治疗对改善患者胆道狭窄和胆汁淤积症状有一定帮助,但并不改变患者的远期临床预后。 肝移植手术仍是终末期PSC患者唯一有效的治疗方式。
信源:
https://www.nhc.gov.cn/yzygj/c100068/202507/5b3f41180a42465eb9eec34597bacaf2.shtml
(本网站所有内容,凡注明来源为“医脉通”,版权均归医脉通所有,未经授权,任何媒体、网站或个人不得转载,否则将追究法律责任,授权转载时须注明“来源:医脉通”。本网注明来源为其他媒体的内容为转载,转载仅作观点分享,版权归原作者所有,如有侵犯版权,请及时联系我们。)








