内皮祖细胞(Endothelial progenitor cells,EPCs)是再生医学研究焦点,起源于骨髓等造血细胞,可调控组织再生与血管完整性,其动员机制能促进多器官损伤后血管新生与内皮修复。在慢性肾脏病(CKD)等疾病状态下,EPCs的数量和功能会发生改变,因此循环EPCs的数量及功能被建议作为评估血管健康的指标,促进EPCs动员或数量增加可能是一种潜在疗法。
近期,《JASN》杂志发布了一篇综述“Endothelial Progenitor Cells: Disease Markers and Potential Therapy in Kidney Disease.”(IF=9.4),全面阐述了当前EPCs在机体稳态中的作用,以及其调控肾脏疾病的相关研究与最新进展。
血液循环中的EPCs及驻留在组织微环境中的EPCs通过细胞替代或释放血管生成物质[包括
EPCs在外周血单核细胞中占比仅0.01-0.3%,为应对血管损伤,需从骨髓动员或采集移植。循环EPCs难以直接从外周血中获取,可从分离的外周血单个核细胞(PBMCs)中培养并扩增内皮细胞,这些细胞与EPCs功能相似,可用于研究其功能及治疗血管损伤。体外培养产生的细胞群体至少分为“早期”(4-7天)和“晚期”(17-23天)群体,还有24-30天的极晚期群体。尽管这些细胞群体功能相同,但其作用模式和机制不同,治疗应用也存在差异。与早期细胞相比,晚期细胞血管生成潜力更强,可直接促进血管生成和修复,对
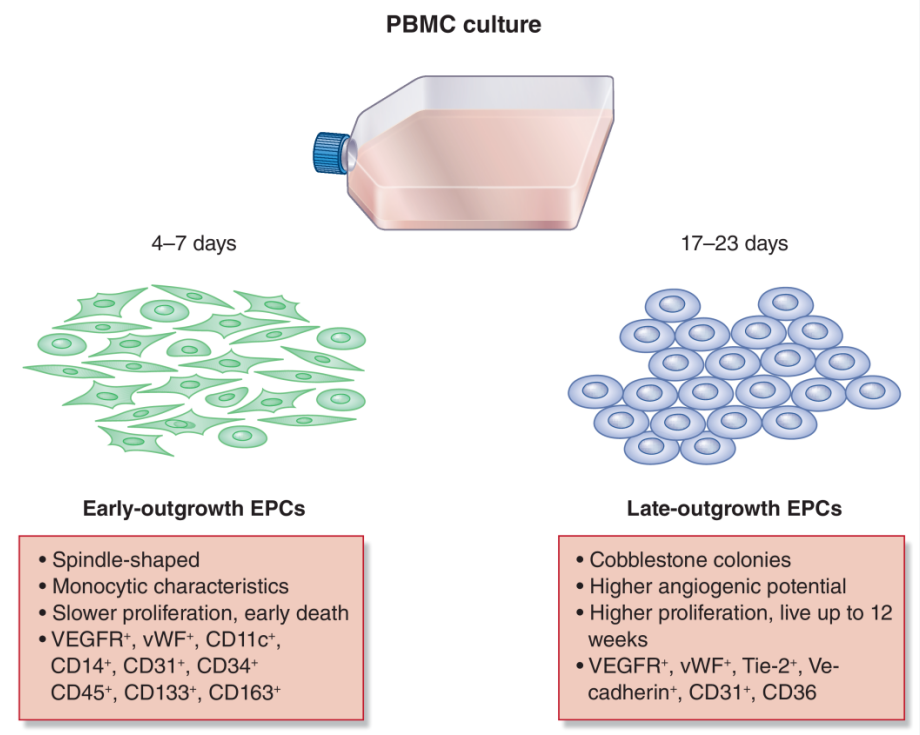
图1. 不同培养时间对内皮祖细胞(EPCs)的类型和形态影响
EPCs的增殖特性有助于其修复和替代靶组织中受损的内皮细胞(图2)。EPCs通过归巢至血管损伤部位,随后分化为成熟内皮细胞、促进裸露血管的再生并抑制细胞凋亡来应对血管损伤,其功能通过旁分泌/自分泌机制实现,释放的微粒或因子可诱导EPCs从骨髓释放、触发内皮细胞增殖,并通过PI3K/AKT通路促进血管生成,在肾脏中可能抑制肾小球硬化和肾小管间质纤维化。
EPCs还通过旁分泌信号和维持血管微环境以支持骨髓微环境。EPCs位于血管周围区域,分可泌基质细胞衍生因子1(SDF-1)、VEGF等关键因子调控造血干细胞的静止、存活、增殖和动员。EPCs不仅在稳态条件下促进造血干细胞在骨髓中的驻留,还能在应激或损伤时调节其动员,这种交互作用对造血稳态和有效的骨髓再生至关重要。自分泌信号通过VEGF等生长因子参与EPCs自我更新和增殖,介导其迁移、归巢和血管生成活性。
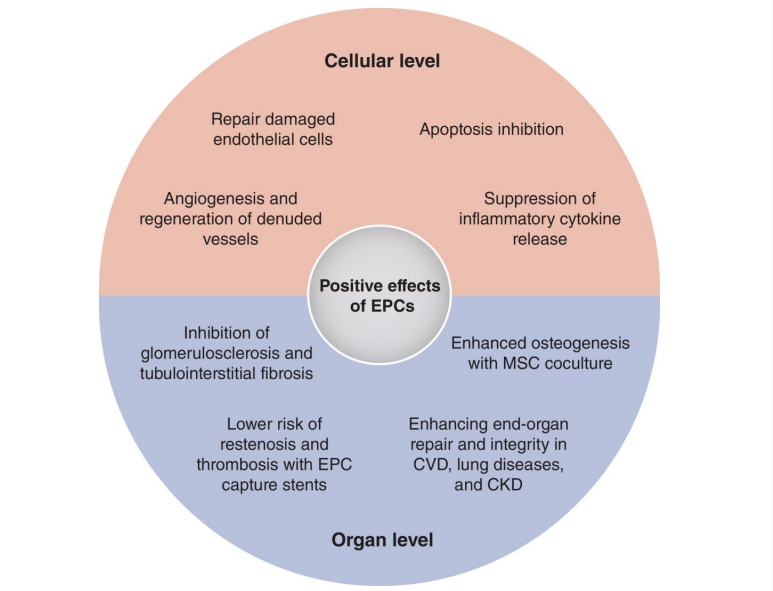
图2. 内皮祖细胞的益处包括血管生成、抗炎和组织修复
这些作用可在细胞水平观察到并转化为器官修复。CVD:心血管疾病;CKD:
特征性表面标志物使EPCs成为评估血管健康与疾病状态的生物标志物。其属造血干细胞谱系,表达CD34因子,呈现血管内皮生长因子受体2(VEGFR-2)阳性,还可能表达CD133。当EPCs向内皮细胞谱系分化时会丢失CD133,开始表达CD31、血管内皮钙黏蛋白和血管性血友病因子,CD31从早期EPCs持续表达至成熟内皮细胞,晚期EPCs强表达CD31等,随成熟逐渐丢失CD133和CD34。
EPCs的动员由SDF-1及其表面表达的趋化因子CXC基序受体(CXCR)4或CXCR7介导。SDF-1从受损器官释放后,与黏附分子相互作用,增强EPC对裸露上皮的附着,从而触发新生血管形成。动员也可能通过一氧化氮(NO)实现:内皮型NO合酶(eNOS)激活后生成NO,可动员EPC、维持稳态并调节血管张力。
心血管疾病和肾脏疾病的主要危险因素包括肥胖和代谢功能障碍。EPCs数量变化机制可能能涉及造血功能的刺激、对血管损伤作出反应的骨髓动员,或髓外脂肪储存库释放EPCs。EPCs发挥血管保护作用的能力不仅取决于其绝对数量,还与其功能特性相关。部分研究指出肥胖可能减少EPCs数量,进而损害内皮修复功能。糖尿病患者的EPCs数量同样减少,尤其是在合并大血管并发症时,EPCs向受损内皮的迁移和归巢功能受损。高血压常损害EPCs的功能和数量,接受药物治疗的高血压患者早期EPCs功能尚可保留,这是因为血管紧张素II可短暂增强EPCs功能,而部分肾素抑制剂会减少早期动脉粥样硬化患者的循环EPCs数量。原发性醛固酮增多症的高血压小鼠表现出EPCs再生与修复失衡。此外,研究表明吸烟也会减少循环EPCs数量。
AKI可导致肾小管上皮细胞损伤和微血管内皮细胞功能障碍(图3)。急性缺血状态会引起含氮废物潴留、血尿酸水平升高,释放血管生成素-2、血管性血友病因子等介质,促使EPCs归巢损伤肾脏。因此,AKI早期EPCs数量增多,但全身炎症、氧化应激和内皮损伤等致EPCs功能受损,迁移、黏附等能力下降,阻碍血管修复。
在临床前AKI模型中,动员或增强EPCs功能的疗法已显示出前景。具有修复作用的EPCs携带miR-126等分子,可保护肾脏血流动力学、减少淋巴细胞浸润。AKI动物模型验证,EPCs治疗可改善IRI小鼠肾功能,iEPCs疗法能预防AKI,为EPCs预防AKI研究开辟道路。
多数研究显示,无论病因如何,CKD患者循环EPCs数量及增殖能力均下降,合并糖尿病者更显著。此外,CKD中EPCs减少还可能继发于需求增加或动员障碍(图3)。一项研究对不同CKD分期患者的循环EPCs类型(CD31/CD34⁺、CD62E/CD34⁺、KDR/CD34⁺、CXCR4/CD34⁺)进行了分类,发现与CKD早期阶段相比,CKD IV-V 期患者的CXCR4/CD34⁺ EPCs数量更少,而SDF-1α和VEGF水平更高。此外,多项研究表明,CKD患者EPCs数量较健康人减少40%,集落数量减少75%。接受血透的儿童EPCs水平降低47%,未透析的CKD儿童则无此现象,提示儿童可能在全身环境中度受损时维持内源性修复系统。
除了减少EPCs数量以外,CKD还会影响EPCs质量:其迁移功能和黏附能力下降30%,干扰血管修复。CKD患者循环CD34⁺ EPCs数量减少且功能受损,随疾病进展而恶化,透析仅能部分改善。
促进肾脏再生的研究已日益受到关注。在小鼠CKD模型中,EPCs治疗可阻止肾功能恶化、减少蛋白尿并维持肾脏结构。人脐带来源的CD34⁺细胞可减轻小鼠肾小管间质损伤并保护微血管。在肾血管疾病猪模型中,自体EPCs可改善肾功能、减轻结构性损伤,并降低心肌损伤。这些研究结果表明,EPCs可能成为进展性CKD及其并发症的潜在治疗手段。
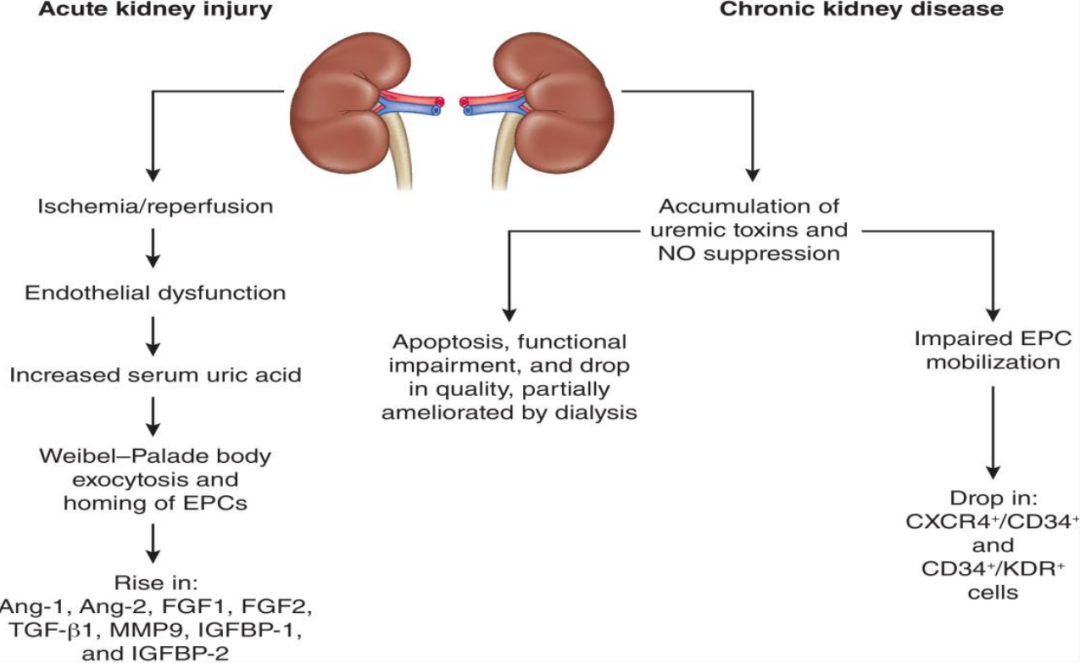
图3. 急性肾损伤和慢性肾脏病中内皮祖细胞的功能变化
多项试验证实,外源性递送EPCs或其旁分泌载体(EPCs来源的细胞外囊泡)可增强心血管保护作用。自体EPCs可抗炎症和/ 或脓毒症,还能增强EPCs归巢并促进组织上皮化,其细胞外囊泡可抑制炎症因子释放,故细胞外囊泡可作EPCs功能替代物。
内源性EPCs数量也可通过干预扩增。SDF-1α通过EPCs表面表达的同源CXCR4受体诱导EPCs动员和归巢,SDF-1α/CXCR4轴可促进内皮细胞中VEGF的表达及Akt磷酸化,这是EPCs归巢和跨内皮迁移至缺血区域的关键通路,SDF-1α肽类似物和CXCR4激动剂CTCE-0214还可通过减少TNF-α释放和上调PI3K-AKT通路来抑制炎症反应。
升高循环EPCs水平的方法还有使用药物(如血管紧张素转换酶抑制剂、他汀类药物、PPAR激动剂、钠-葡萄糖协同转运蛋白2抑制剂)、体育锻炼和缺血预处理等,其作用机制包括抑制NF-κB或增加NO生成,微重力环境也可通过上调HIF-1α和促进NO释放(图4)。除增加EPCs循环水平外,靶向EPCs至特定器官的策略已开发。如在心肌缺血小鼠模型中,将中性粒细胞负载与EPCs表面标志物CD34抗体偶联的超顺磁性氧化铁纳米颗粒,经全身给药后,EPCs向损伤部位的募集增加了三倍,心功能得到改善。
EPCs可作为辅助治疗手段,与间充质干细胞共培养时释放可溶性因子,改善骨血管化并协同促进血管生成与骨生成。此外,包被CD34+抗体的EPCs捕获支架能促进内皮化、降低血栓风险,但因晚期管腔丢失等问题限制应用。低能量冲击波疗法(LESW)可诱导归巢因子释放,提升循环EPCs水平,增强其迁移与存活能力,在猪肾血管疾病模型中证实能改善EPCs动员及肾脏功能。两项针对猪肾动脉狭窄血运重建的研究探索了EPCs作为辅助治疗的潜力,发现与单纯血运重建相比,肾动脉内联合递送自体EPCs可改善肾脏预后,显著促进肾脏结构与功能的保护,为肾血管疾病所致肾损伤的治疗提供了新途径。
尽管EPCs通常可维持血管健康和体内稳态,但某些亚型如CD133+ EPCs可加剧AKI、导致内皮功能障碍,OC+EPCs可促进动脉粥样硬化,急性肺损伤中EPCs可能因输注毒性等引发不良反应。外源性EPCs临床应用受限,目前大多数研究仅通过EPCs评估疾病严重程度,仅少数临床试验将其作为肾脏疾病主要治疗手段。
总结
内皮祖细胞作为评估心血管与肾脏健康的重要生物标志物,兼具显著的治疗潜力。越来越多的研究证实了其在再生医学领域的多样化功能、细胞类型及应用价值。同时辅助治疗和增强EPCs动员技术的持续进展,为改善肾脏血管和组织修复策略提供了新途径。未来需通过更多研究探索其临床转化路径,推动这一潜在治疗工具在肾脏修复中的实际应用。

(本网站所有内容,凡注明来源为“医脉通”,版权均归医脉通所有,未经授权,任何媒体、网站或个人不得转载,否则将追究法律责任,授权转载时须注明“来源:医脉通”。本网注明来源为其他媒体的内容为转载,转载仅作观点分享,版权归原作者所有,如有侵犯版权,请及时联系我们。)








